
Cet article de revue fait la synthèse des recherches en paléogénomique couvrant les temps très anciens du Pléistocène, non seulement supérieur, mais aussi moyen et inférieur. Le Pléistocène est la période géologique qui précède l'Holocène, période que nous traversons actuellement et qui correspond à la période interglaciaire actuelle. Le Pléistocène lui, correspond aux diverses périodes glaciaires allant de 2,6 millions d'années (2,6 Ma) à 11700 ans (11,7 ka) avant le présent, c'est à dire jusqu'au début du néolithique.
Les différentes périodes du Pléistocène se décomposent en :
- Pléistocène inférieur (2,58 Ma à 781 000 ans avant le présent) subdivisé en deux étages géologiques
- Gélasien (2,58 à 1,80 Ma avant le présent),
- Calabrien (1,80 Ma à 781 000 ans avant le présent) ;
- Pléistocène moyen ou « Ionien » (781 000 à 126 000 ans avant le présent) ;
- Pléistocène supérieur ou « Tarentien » (126 000 à 11 700 ans avant le présent).
L'ADN est d'autant plus dégradé que les fossiles desquels ils sont prélevés sont âgés. Svante Pååbo a été un pionnier dans l'étude des ADN anciens et en développant des méthodes pour les différencier des contaminations, non seulement dues à l'expérimentateur, mais aussi aux végétaux et bactéries qui l'ont contaminés au cours des millénaires successifs. Il a ainsi, en réunissant l'information obtenue à partir de nombreux fragments d'ADN, reconstitué tout le génome de l'Homme de Néandertal et également découvert qu'il avait existé une autre espèce d'humain, contemporain à Néandertal, qu'il a appelé l'Homme de Denisova. Ses travaux ont été couronnés par l'attribution du Prix Nobel de Physiologie ou Médecine 2022.
Toutefois, les travaux de l'équipe de Pååbo couvrait essentiellement des fossiles de Neandertal datant du Pléistocène supérieur. L'article de revue ci-dessous fait le point sur les travaux qui ont déjà pu être entrepris sur des espèces ayant vécu en des âges beaucoup plus reculés des Pléistocènes moyen et inférieur. Les auteurs expliquent également les mécanismes de dégradation de l'ADN, comment ils compliquent la tâche, mais comment aussi on peut en bénéficier pour distinguer un ADN très ancien de contaminations plus récentes. Enfin, ils terminent par des perspectives sur les recherches futures, et comment les résultats attendus pourraient être exploités pour comprendre l'évolution actuelle de l'Homme et des autres espèces animales.
Paléogénomique des périodes très anciennes et limites de la survie de l’ADN
Traduit de : Love Dalén, Peter D. Heintzman, Joshua D. Kapp, Beth Shapiro, SCIENCE, 5 Oct 2023, Vol 382, Issue 6666 pp. 48-53
Résumé
Bien que la plupart des études anciennes sur l’ADN se soient concentrées sur les 50 000 dernières années, les approches paléogénomiques peuvent désormais atteindre le début du Pléistocène, une époque de changements environnementaux répétés qui ont façonné la biodiversité actuelle. Les transects génomiques émergents des temps reculés, notamment à partir de l'ADN conservé dans les sédiments, permettront de déduire une évolution adaptative, la découverte d'espèces non reconnues et l'exploration de la façon dont les glaciations, le volcanisme et les inversions paléomagnétiques ont façonné la démographie et la composition des communautés. Dans cette revue, nous explorons ce qu’on arrive à faire aujourd’hui’hui en paléogénomique et discutons des principaux défis, notamment les limitations techniques, les divergences évolutives et les biais associés, ainsi que la nécessité d'une datation plus précise des restes et des sédiments. Nous concluons qu’en améliorant les méthodes de laboratoire et informatiques, le domaine émergent de la paléogénomique des époques très lointaines élargira l'éventail des questions pouvant être résolues à l'aide de l'ADN ancien.
L’époque du Pléistocène [ il y a environ de 2,6 millions d’années (Ma) à 11 700 ans (ka) ] a été une période de bouleversements environnementaux considérables qui ont façonné la répartition mondiale actuelle de la biodiversité. Les changements environnementaux au cours du Pléistocène comprenaient des fluctuations cycliques des températures mondiales et des régimes de précipitations, des avancées et des récessions des calottes glaciaires des hautes latitudes et des changements substantiels du niveau de la mer, ainsi qu'un volcanisme à grande échelle, des inversions paléomagnétiques et la propagation mondiale de l'homme (1). Ces événements ont modifié les habitats du monde entier, entraînant des changements dans la disponibilité des ressources et la composition des communautés écologiques.
Les riches archives fossiles du Pléistocène ont joué un rôle déterminant pour tester les hypothèses sur la corrélation entre ces changements environnementaux et la dynamique de la biodiversité, en particulier aux hautes latitudes où le climat froid favorise la préservation des fossiles. Cela est particulièrement vrai pour le Pléistocène supérieur (126 à 11,7 ka), grâce aux inférences très fines permises par l'ADN ancien conservé dans des fossiles datant de cette période. De telles inférences ont permis de mieux comprendre le renouvellement de la population (2, 3, 4) et le flux génétique interspécifique (5) – processus invisibles pour les techniques paléontologiques traditionnelles – et ont montré que les tendances démographiques des grands mammifères suivent de près l’habitat disponible (6).
Les progrès techniques dans la récupération de l’ADN ont étendu la capacité de faire ces déductions en avançant plus profondément dans le Pléistocène. L’ADN d’os et de dents vieux de plusieurs centaines de milliers d’années (7, 8, 9) et de plus d’un million d’années (10) a désormais été récupéré et analysé (Fig. 1). De tels paléogénomes très anciens, que nous considérons ici comme faisant référence à des génomes assemblés à partir d'organismes ayant vécu pendant ou avant le Pléistocène moyen, c'est-à-dire > 126 ka, sont encore rares car les processus post-mortem conduisent à une dégradation des molécules d'ADN en fragments de plus en plus petits, ce qui rend la récupération de l'ADN plus difficile avec l'âge. L'ADN du Pléistocène inférieur et moyen a cependant été récupéré à partir de restes et de sédiments dans le pergélisol des hautes latitudes (10, 11, 12, 14) et dans les grottes des basses latitudes (15, 16), ce qui suggère que la génomique de ces temps très reculés est réalisable dans des environnements de préservation idéaux. Nous explorons ici l’état actuel de la recherche en paléogénomique de ces temps reculés, les principaux obstacles empêchant une adoption plus large et les questions scientifiques que la paléogénomique de ces temps très anciens peut résoudre.
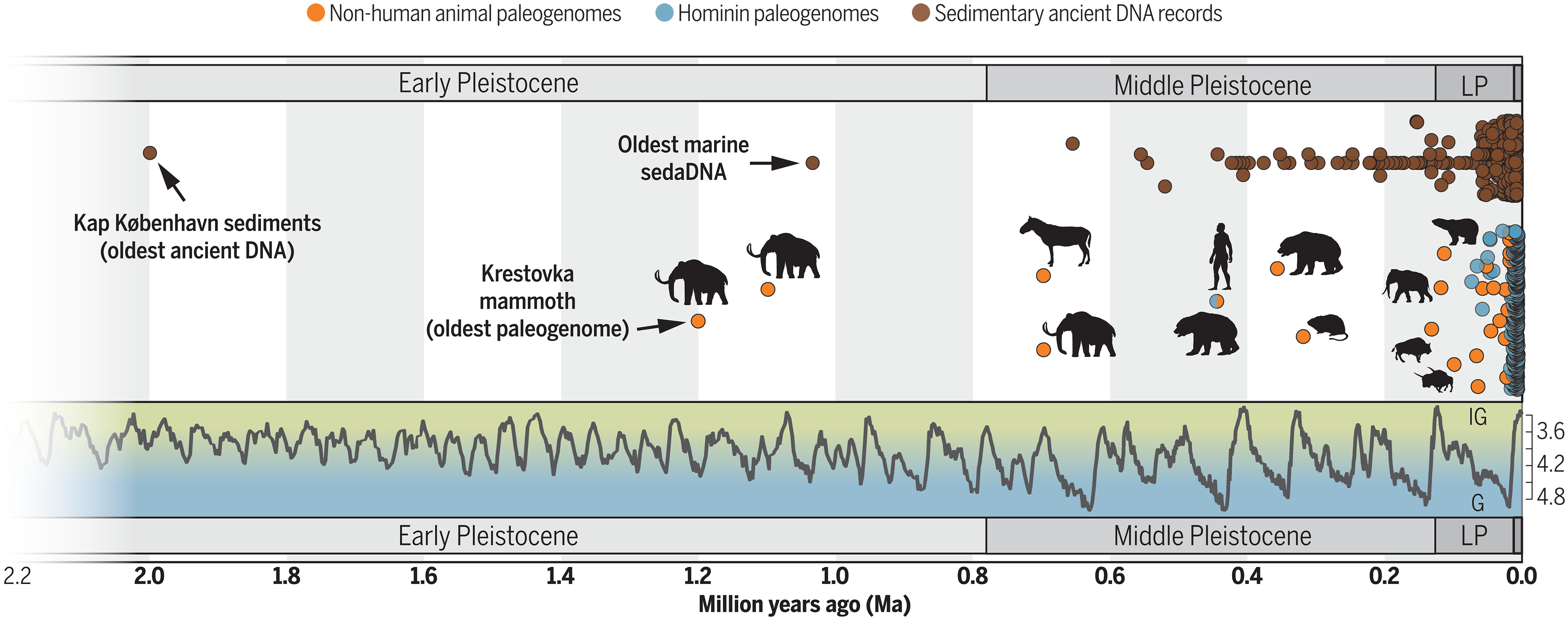
Fig. 1. La répartition temporelle des études sur l'ADN ancien à ce jour met en évidence les lacunes et les opportunités pour la paléogénomique des temps reculés et l'ADN ancien sédimentaire.
La plupart des études d’ADN anciens datent des 50 derniers ka et du cycle glaciaire le plus récent. La courbe climatique est basée sur les mesures de l'oxygène δ18 benthique [ par mil ; Pile LR04 de (42) ]. Les données d'ADN ancien sédimentaire proviennent de AncientMetagenomeDir (v23.06.0) (57) et (58), les enregistrements de métabarcodes datant de plus d'un million d'années étant exclus. Les données paléogénomiques sont disponibles auprès de (59). Les paléogénomes âgés de plus de 100 ka sont annotés d'une silhouette du taxon étudié, les paléogénomes des temps très anciens comprenant un bison des steppes de 130 ka (36) ; Lemming à collier de 330 ka (40) ; Ours des cavernes de 360 ka (9) ; Ours des cavernes et hominidés de 430 ka (35, 60) ; Cheval de 700 ka (8) ; et mammouths de 700 ka, 1,1-Ma et 1,2-Ma (10). LP, Pléistocène supérieur ; IG, Interglaciaire ; G, Glaciaire. Les silhouettes proviennent de PhyloPic https://beta.phylopic.org/ , sont dans le domaine public et sont à créditer à Zimices (mammouth, deux bisons) et Robert Bruce Horsfall (cheval)
Persistance de l'ADN dans les temps très anciens
L’ADN ne survit pas indéfiniment, mais il survit beaucoup plus longtemps que ne le prédisaient les premiers modèles. En 1993, Lindahl estimait que la dépurination hydrolytique conduirait à une dégradation complète des molécules d'ADN en plusieurs dizaines de milliers d'années (17). Cette limite a depuis été dépassée et l'ADN est régulièrement récupéré à partir de restes et de sédiments datant des 100 derniers ka. En septembre 2023, le paléogénome reconstruit le plus ancien provenait d'un mammouth préservé dans le pergélisol datant d'entre 1 et 2 Ma (10) et l'ADN isolé le plus ancien provenait d'un sédiment d'environ 2 Ma du nord du Groenland (11). Cependant, l’âge maximum des molécules d’ADN récupérables et utiles – celles qui sont suffisamment longues pour conserver des informations – reste incertain.
L'ADN commence à se dégrader immédiatement après la mort de l'organisme, initialement par l'activité des nucléases microbiennes et endogènes (Fig. 2). Dans l’ADN nucléaire, les brins sont clivés dans les régions labiles des complexes histones-ADN, ce qui entraîne une périodicité d’environ 10 bases dans la distribution des longueurs des molécules récupérées (18). Le principal mécanisme chimique de fragmentation de l’ADN est la dépurination hydrolytique. Ce processus élimine les bases adénine ou guanine, créant des sites abasiques qui peuvent être clivés par élimination β (19) (Fig. 2C) et conduisant à une surreprésentation des purines au niveau des cassures de brins (20) (Fig. 2E) et des vides intérieurs (21). La désamination hydrolytique, une autre forme courante de dommage chimique, convertit la cytosine en uracile et est observée sous forme de thymine dans les données de séquençage, ou « transitions C-to-T » (Fig. 2C). La désamination se produit principalement près des extrémités des brins et dans l'ADN simple brin (17, 21, 22) (Fig. 2E). La réticulation de l'ADN (19, 22) et les dommages oxydatifs (20, 23) se produisent également, mais sont observés moins fréquemment que la dépurination et la désamination. Ces modèles de dommages typiques peuvent être utilisés pour corroborer bioinformatiquement l’authenticité des séquences anciennes récupérées et, pour réduire leur impact sur la précision des séquences, elles peuvent être identifiées et supprimées des ensembles de données d’ADN anciens à l’aide d’approches bioinformatiques standard.
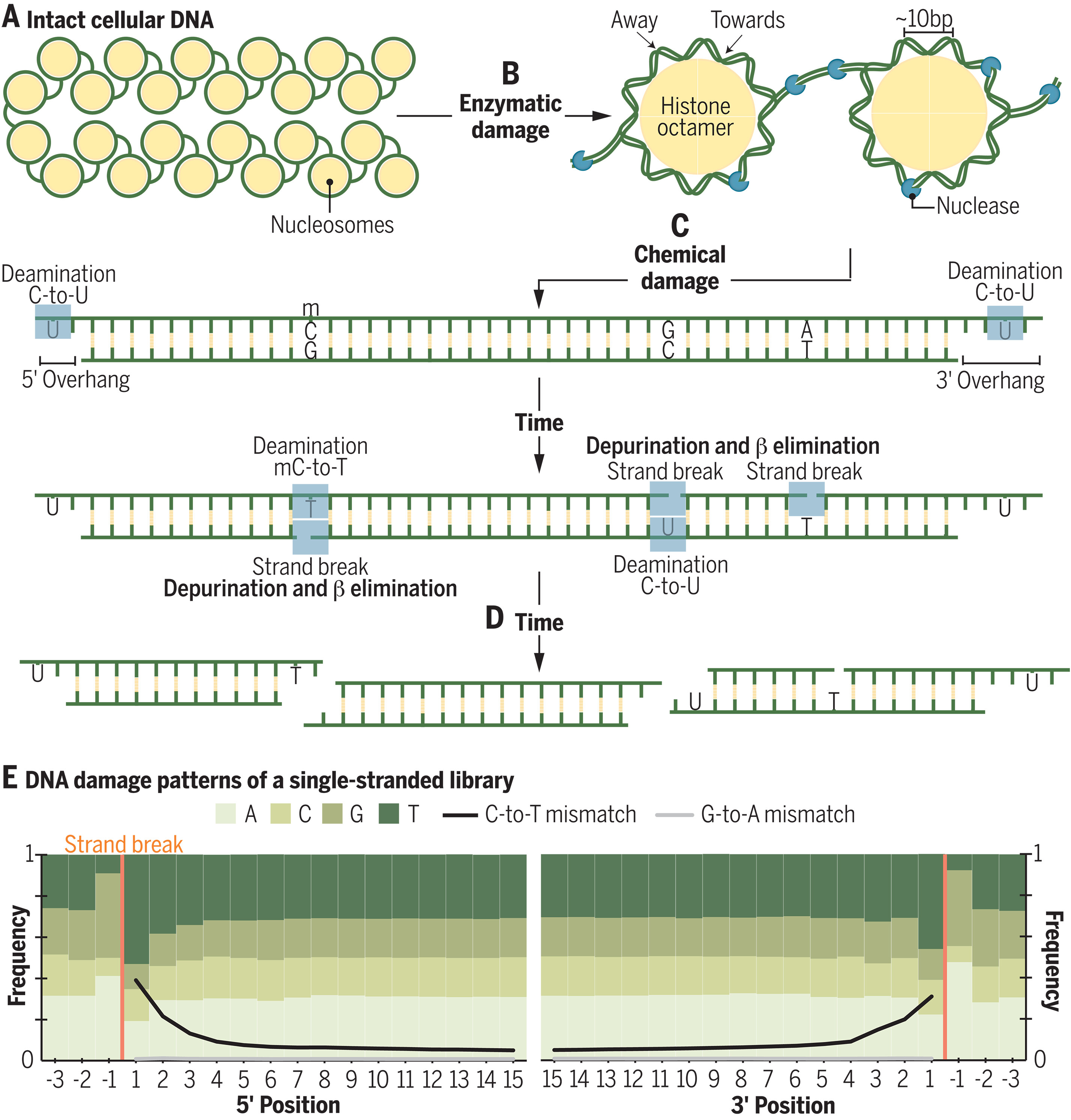
Fig. 2. La fragmentation et la dégradation de l'ADN commencent après la mort et se poursuivent jusqu'à ce que les fragments soient trop courts pour être utiles.
(B) Après la mort, la réparation s’arrête et les dommages à l’ADN commencent à s’accumuler. Les nucléases et les micro-organismes clivent l'ADN dans les régions labiles entre les nucléosomes et lorsque le squelette de l'ADN s'éloigne des histones.
(C) Au fil du temps, les dommages chimiques s’accumulent également. Les cytosines sont converties en uraciles et les cytosines méthylées sont converties en thymines (par désamination). Les cytosines sont particulièrement vulnérables à la désamination dans les régions simple brin telles que les régions pendant aux extrémités de l'ADN, mais la désamination est possible dans certains contextes double brin. La fragmentation se produit après la perte des bases puriques (dépurination), créant des sites abasiques qui peuvent être clivés par élimination β. La dépurination et l'élimination β créent une région d'ADN simple brin, qui laisse les cytosines vulnérables à la désamination.
(D) Après suffisamment de temps, les molécules d’ADN deviendront trop courtes pour être identifiables.
(E) Un résumé des fréquences de base et de mésappariement le long des 15 bases initiales 5 'et 3' des lectures générées à l'aide d'un protocole de bibliothèque d'ADN simple brin [bibliothèque JKFC14 ; (25)]. La dépurination conduit à une surreprésentation des bases adénine et guanine adjacentes aux cassures de brins. Les inadéquations C-to-T sont élevées près des extrémités de lecture et observées tout au long des lectures endommagées. Alors que des mésappariements 3’ G-A sont observés dans les bibliothèques double brin, les bibliothèques simple brin présentent un signal C vers T aux deux extrémités en conservant les extrémités natives des molécules.
La récupération d’ADN de plus en plus vieux et endommagé est possible en partie grâce aux progrès techniques réalisés en laboratoire. Les anciennes méthodes d’isolement de l’ADN sont optimisées pour récupérer à la fois les molécules d’ADN courtes et les molécules contenant des coupures et des lacunes. Les molécules extraites sont préparées pour le séquençage en attachant des adaptateurs spécifiques à de l'ADN double brin ou à du simple brin. Les approches simple brin de préparation de bibliothèques génomiques (24, 25) convertissent de manière native l'ADN simple brin ainsi que l'ADN double brin et convertissent plus efficacement les molécules contenant des coupures et des lacunes par rapport aux approches double brin. Les extraits d'ADN sont également souvent traités avec l'ADN glycosylase et l'endonucléase VIII de l'uracile pour réduire les dommages causés par la désamination en éliminant les bases uracile (26). Bien que cette approche réduise les erreurs induites par les dommages dans les données de séquençage résultantes, elle coupe également le squelette de l'ADN au niveau des sites abasiques et raccourcit les molécules récupérées de 5 à 10 nucléotides (26). Les molécules d’ADN des temps très anciens sont déjà courtes – souvent < 35 bases (15) – et par conséquent cela peut réduire la proportion d’ADN endogène utile.
La nature courte de ces molécules d’ADN les rend sujettes à un alignement parasite et à des biais (27), compliquant l’assemblage et l’analyse du génome. Par exemple, les anciens ensembles de données sur l’ADN comprennent à la fois l’ADN endogène des organismes cibles et l’ADN exogène introduit. Ces catégories de molécules peuvent être séparées en identifiant chaque lecture via une affectation taxonomique, ce qui peut être problématique si l'organisme ancien n'a pas de parent vivant proche pour servir de référence génomique. L’absence de référence étroite, les biais de référence et les erreurs introduites par les dommages entraveront également la distinction de variantes et l’obtention d'un consensus dans l’interprétation. Les approches bioinformatiques atténuent ces défis en modélisant directement les dommages et/ou les biais de l'ADN dans le cadre du génotypage (28) ou en considérant uniquement les substitutions qui ne sont pas affectées par la désamination de la cytosine. Les génomes de référence peuvent également être modifiés pour créer des références artificiellement plus proches, comme une version « néanderthalisée » du génome humain de référence pour servir de référence à la cartographie aux lectures de génomes néandertaliens (29). Des génotypes vraisemblables plutôt que des génotypes stricto-senso peuvent également être utilisés lors de l'analyse en aval, bien que les méthodes analytiques basées sur l'imputation puissent être inappropriées pour les ensembles de données des temps très anciens si la diversité génomique ancienne n'est pas représentée dans les panels de référence existants.
Opportunités de recherche découlant de l’ADN des temps très anciens
Différentiation des espèces et évolution
La différentiation des espèces n'est pas toujours un simple processus de cladogenèse suivi d'un isolement reproductif. Au lieu de cela, les données modernes et paléogénomiques ont montré que l'hybridation interspécifique est étonnamment courante et peut-être due en partie à une redistribution répétée de l'habitat associée aux cycles glaciaires (5, 9, 10). Par exemple, les ours bruns et les ours polaires se sont hybridés au cours des périodes glaciaires et interglaciaires précédentes (30, 31) ainsi qu'à l'ère moderne. Récemment, des paléogénomes d’ours polaires et d’ours des cavernes datant de 360 ka ont révélé que tous les ours bruns vivants tirent une partie de leur ascendance d’un mélange avec ces autres lignées d’ours – des événements évolutifs qui étaient invisibles sans ces paléogénomes (9, 32). De même, un paléogénome de mammouth datant du Pléistocène inférieur a révélé que les mammouths colombiens (Mammuthus columbi) sont nés d'une hybridation entre deux anciennes lignées de mammouths distinctes (10) (Fig. 3). Une diversité taxonomique des paléogénomes anciens pourraient clarifier le moment, le taux et l’étendue des épisodes d’introgression génomique ainsi que leur rôle dans l’évolution. Les données paléogénomiques d'espèces disparues au cours du Pléistocène inférieur et moyen, telles que les hyènes à face courte, les jaguars européens et les énigmatiques canidés Xenocyon, pourraient permettre de déterminer si ces taxons ont contribué à la constitution génétique des carnivores vivants. Les paléogénomes anciens pourraient également identifier des lignées « fantômes » inconnues qui ont contribué aux ancêtres des espèces, comme l’illustre la caractérisation paléogénomique du mammouth de Krestovka (10) (Encadré 1 et Fig. 3).
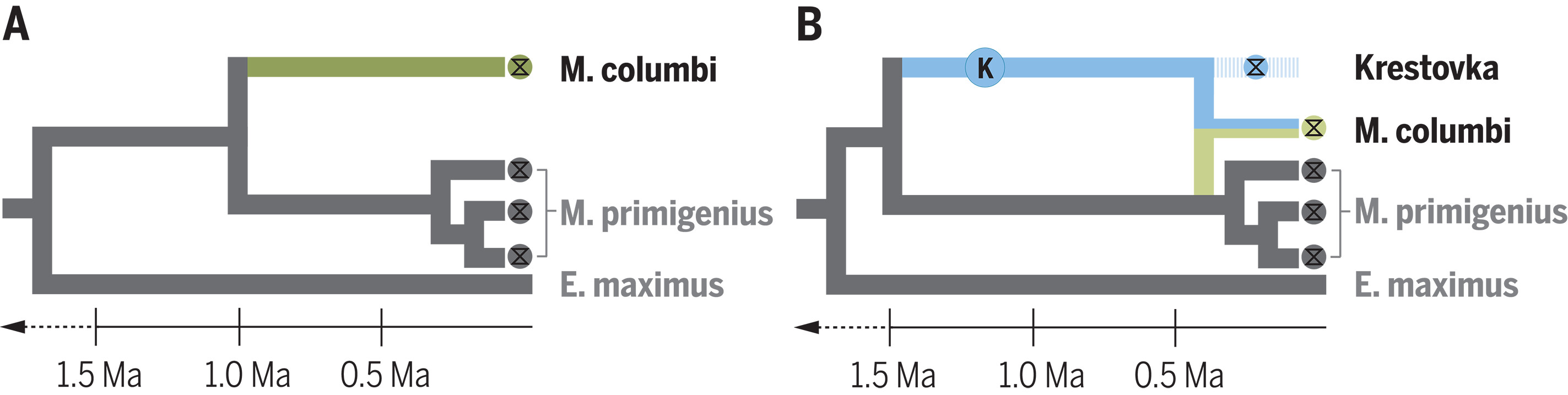
Fig. 3. Les paléogénomes des temps très anciens ont permis de mieux comprendre l'histoire évolutive des mammouths.
(A) Les hypothèses paléontologiques supposaient que la lignée de M. columbi avait évolué après une divergence précoce avec M. primigenius.
(B) Cependant, l'isolement d'un paléogénome profond du mammouth de Krestovka (cercle bleu) a révélé que M. columbi a émergé plus récemment suite à un mélange entre le Lignées Krestovka et M. primigenius.
Adapté de (10).
ENCADRÉ 1
ADN de mammouth des temps très anciens et inférence de toute l’histoire évolutive d’une lignée.
La puissance d'une approche génomique en profondeur a été mise en évidence dans une étude menée par van der Valk et ses collègues (10) dans laquelle des données pangénomiques ont été collectées auprès de trois mammouths de Sibérie datés d'environ 700 ka à 1,2 Ma, ce qui a permis d'examiner l’évolution des mammouths sous plusieurs angles génomiques :
Une nouvelle lignée :
Le plus ancien des spécimens de mammouth appartenait à une lignée évolutive jusque-là inconnue et divergente, Krestovka. Cela impliquait que deux lignées distinctes de mammouths, Krestovka et les ancêtres des mammouths laineux, vivaient en Sibérie au cours des derniers stades du Pléistocène inférieur. L'analyse a également révélé que les mammouths appartenant à la lignée Krestovka ont été les premiers mammouths à coloniser l'Amérique du Nord il y a 1,5 à 1,2 Ma (61).
Hybridation :
Plusieurs éléments de preuve suggèrent que le mammouth colombien est né d'une hybridation entre la lignée Krestovka et les premiers mammouths laineux. Cette hybridation a eu lieu lorsque les mammouths laineux se sont développés en Amérique du Nord au cours du Pléistocène moyen, après que la lignée Krestovka était déjà établie sur le continent (Fig. 3). Les mammouths colombiens tirent environ 50 % d'ascendance de chacune de ces deux lignées.
Évolution adaptative :
La nature très ancienne du gigantesque ensemble de données a permis à van der Valk et al. d’estimer le taux d'évolution adaptative chez les mammouths. Ils ont conclu que l’origine de la lignée du mammouth laineux ne coïncidait pas avec un taux accru de changements codant pour des protéines et donc à un taux plus élevés de sélection positive dans le génome (10). Au contraire, des analyses ultérieures ont identifié une suite de gènes qui ont subi des modifications codant pour des protéines au cours des 700 derniers ka et étaient donc spécifiques aux mammouths laineux (37).
L’ADN très ancien peut révéler des instantanés génomiques de l’histoire évolutive complète d’une espèce individuelle (Encadré 1). Comme de nombreux oiseaux et mammifères adaptés aux climats tempérés et froids font remonter leur origine au Pléistocène inférieur et moyen (33, 34), les paléogénomes de ces espèces pourraient corréler les changements évolutifs à des perturbations environnementales spécifiques, telles que les transitions entre les régimes climatiques ou le remaniement des communautés. Le processus de spéciation (différentiation des espèces) peut être étudié au fur et à mesure, en explorant les goulots d'étranglement des événements fondateurs et en vérifiant si la spéciation s'est produite par allopatrie stricte ou progressivement avec un flux génétique post-divergence. Comme les paléogénomes des temps anciens ont tendance à occuper des positions phylogénétiques basales au sein de leurs clades, ils peuvent également fournir des étalonnages importants pour estimer les taux d’évolution moléculaire. Par exemple, les données paléogénomiques d'un hominidé du Pléistocène moyen de Sima de los Huesos dans l'Espagne actuelle ont confirmé les hypothèses génomiques du Pléistocène supérieur selon lesquelles les Néandertaliens et les Dénisoviens ont divergé au début du Pléistocène moyen (35), alors que l'inclusion d'un paléogénome de cheval d'environ 700 ka dans la phylogénie des équidés a reculé l’origine estimée des équidés vivants à plus de deux fois celle supposée précédemment (8).
Les paléogénomes du Pléistocène inférieur et moyen peuvent également être utilisés pour tester des hypothèses sur les relations entre les espèces, notamment sur la manière dont les formes dérivées sont liées aux formes antérieures. Une question en suspens en paléontologie est de savoir si les morphoespèces fossiles sont de véritables espèces, des écomorphes synchrones ou des chrono-espèces qui étaient les ancêtres directs des espèces successives. Une étude paléogénomique d'anciens bisons d'Amérique du Nord datant d'environ 130 à 110 ka, par exemple, a montré que deux échantillons présentant un dimorphisme de taille extrême et représentant des espèces soi-disant distinctes – le bison de Longhorn et le bison des steppes – appartiennent en réalité à la même lignée qui s'est dispersée dans l’Amérique du Nord seulement quelques dizaines de milliers d’années plus tôt (36). À l’inverse, la paléogénomique des temps reculés peut également donner un contexte à des espèces pour lesquelles nous ne disposons que des restes limités, comme les Dénisoviens (35).
Enfin, les paléogénomes à des échelles de temps très lointains permettront d'explorer les aspects de l'évolution adaptative. Au niveau le plus élémentaire, les génomes très anciens peuvent aider à identifier le moment où des mutations adaptatives sont apparues. Par exemple, une analyse comparative de paléogénomes de mammouths âgés de quelques milliers à plus d'un million d'années a permis d’identifier des gènes associés au développement des cheveux et de la peau, au stockage et au métabolisme des graisses, au fonctionnement du système immunitaire et à la taille du corps qui ont évolué dans la lignée des mammouths laineux durant les derniers 700 ka (37). Les paléogénomes permettront également d'explorer la façon dont le taux de changements du codage des protéines varie au fil du temps, par exemple en conjonction avec les changements climatiques passés, ainsi que d'évaluer le moment où les délétions génomiques sont apparues et le taux de sélection positive et purificatrice dans les régions génomiques introgressées.
L’impact des cycles glaciaires sur la biodiversité
Jusqu'à présent, presque toutes les études sur l'ADN ancien se sont concentrées, pour des raisons pratiques, sur des matériaux du Pléistocène supérieur ou plus récents (Fig. 1). Ainsi, notre compréhension actuelle des processus évolutifs au cours des sous-époques du Pléistocène inférieur (2,6 Ma à 780 ka) et moyen (780 à 126 ka) repose principalement sur des approches plus traditionnelles, notamment la morphométrie, l'analyse des isotopes stables et les enregistrements polliniques. L’accès accru aux données génomiques provenant de fossiles et de sédiments datant de ces sous-époques antérieures permettra de tester plus explicitement les hypothèses sur la manière dont les cycles glaciaires affectent l’évolution et la biodiversité.
Un attribut particulier du Pléistocène antérieur, par exemple, est le changement de périodicité des glaciations de cycles d'environ 40 ka à des cycles d'environ 100 ka survenus entre 1,2 et 0,7 Ma (38) (Fig. 1). Ce changement a entraîné l'isolement des espèces tempérées dans des refuges glaciaires pendant des périodes plus longues, laissant plus de temps pour l'adaptation locale et augmentant le taux de divergence des populations. Les communautés biologiques peuvent également avoir été remaniées, car les glaciations plus longues et de plus grande amplitude ont permis une accumulation suffisante de calotte glaciaire pour la formation du pont terrestre de Béring, rendant possible la dispersion terrestre entre l'Eurasie et l'Amérique du Nord.
Depuis le changement de périodicité glaciaire, le modèle dominant a été des cycles de longues glaciations séparées par des interglaciaires courts et chauds. On pense que cette tendance a influencé la démographie et la dynamique de l’aire de répartition de nombreuses espèces (39). Les longs interglaciaires, par exemple, ont été corrélés à des goulots d'étranglement chez les taxons adaptés au froid (40) et à l'expansion et à la spéciation chez les taxons adaptés au chaud (41). L'interglaciaire inhabituellement long qui s'est produit entre 420 et 370 ka (stade isotopique marin 11) est particulièrement intéressant (42). Les paléogénomes d'individus ayant vécu pendant ce long goulot d'étranglement et avant pourraient être utilisés pour tester ces hypothèses et révéler des changements évolutifs qui ont été écrasés par des goulots d'étranglement génétiques ultérieurs.
Inférence d'écosystèmes anciens
Nous avons décrit des informations qui pourraient être dérivées de l'ADN extrait des restes d'individus ayant vécu au Pléistocène moyen et antérieur. Cependant, les progrès qui permettent la paléogénomique des temps très anciens permettent également de reconstruire des communautés écologiques entières de ces temps reculés. À ce jour, seules cinq études ont tenté d’utiliser l’ADN sédimentaire ancien pour reconstruire des communautés végétales et/ou animales datant du Pléistocène moyen ou plus : Kjær et al. (11) ont reconstruit les composants d'un écosystème interglaciaire du Pléistocène inférieur à partir de sédiments extraits du désert polaire actuel du nord du Groenland ; Armbrecht et coll. (43) ont reconstruit un écosystème marin du Pléistocène inférieur au Pléistocène moyen à partir de l'Iceberg Alley dans l'océan Austral ; Courtin et coll. (12) ont reconstruit un écosystème interglaciaire du Pléistocène moyen à partir d'un méga-effondrement de pergélisol en Sibérie orientale ; et Willerslev et coll. ont reconstruit des communautés végétales du Pléistocène moyen à partir de sédiments collectés sous la calotte glaciaire du Groenland (14) et du pergélisol côtier de Sibérie (13). Parmi ceux-ci, Kjær et al. et Armbrecht et al. ont enrichi les bibliothèques de séquences d'intérêt par hybridation avec des appâts synthétisés conçus pour cibler les taxons arctiques ou antarctiques. Contrairement à l’approche de la PCR basée sur le métabarcodage utilisée par les trois autres études, l’enrichissement ciblé basé sur l’hybridation peut capturer des molécules de n’importe quelle longueur et est donc puissant même lorsque les molécules préservées sont courtes. Bien que l’approche basée sur l’hybridation soit actuellement limitée à la capture de séquences génétiquement similaires à d’autres taxons connus, les améliorations méthodologiques de la capture par hybridation constituent un domaine de recherche mûr qui élargira l’accès à l’ADN sédimentaire très ancien.
La recherche en profondeur sur l'ADN sédimentaire permettra de mieux comprendre l'effet des transitions glaciaires-interglaciaires sur la composition des communautés. Les reconstructions de communautés couvrant la transition vers l’Holocène actuel, par exemple, ont révélé un renouvellement biologique rapide qui suivait de près les changements abiotiques (44, 45). La comparaison avec des transitions plus anciennes permettra de déterminer si les modèles sont prévisibles ou idiosyncratiques, si certaines espèces ou communautés sont plus résiliantes que d’autres aux bouleversements environnementaux, et si certaines transitions ou événements laissent des signatures durables sur la biodiversité des communautés.
Les reconstructions de communautés qui ont prospéré au cours des interglaciaires chauds du passé pourraient fournir un aperçu de la composition potentielle des communautés dans un monde futur plus chaud (11) et améliorer notre compréhension de la façon dont les interactions au niveau des écosystèmes entre les espèces évoluent et se maintiennent. Ils enrichissent également notre compréhension de ces écosystèmes disparus au-delà de ce que l’on peut connaître à partir des archives fossiles. L'ADN sédimentaire profond du nord du Groenland, par exemple, a révélé un l’existence mastodonte ou un animal ressemblant à un mastodonte qui faisait partie de la communauté du Pléistocène inférieur (11), bien qu'aucun reste fossile d'un tel animal n'ait été découvert. L’ADN sédimentaire profond peut également révéler une connectivité passée entre les populations, comme l’indique une étude récente de l’ADN sédimentaire du Pléistocène supérieur provenant d’une grotte au Mexique qui reliait une population éteinte d’ours noirs à des populations vivantes de l’est de l’Amérique du Nord (46). À mesure que les technologies s'améliorent, en particulier celles qui permettent un enrichissement ciblé de plus en plus sensible, nous envisageons l'ADN sédimentaire profond comme un outil puissant pour explorer les conséquences écologiques et évolutives des changements environnementaux sur la biodiversité au niveau communautaire.
Recherches futures pour permettre la récupération de l'ADN des temps très anciens.
Il a été démontré que l’ADN peut survivre dans des conditions de conservation idéales au moins jusqu’au Pléistocène inférieur. La prochaine phase de la recherche sur l’ADN ancien consiste à élargir la gamme taxonomique, géographique et temporelle de l’ADN ancien récupéré et authentifié. Ce défi présente de nouvelles opportunités de recherche sur le terrain, au laboratoire et en bioinformatique.
La génomique des âges reculés est aujourd’hui principalement menée sur des substrats présentant une préservation optimale de l’ADN, tels que ceux issus du permafrost ou des grottes. Cependant, des approches plus efficaces pour récupérer d’anciennes molécules d’ADN continueront d’élargir la gamme d’échantillons et de substrats adaptés à l’analyse. Aujourd’hui, les méthodes d’extraction d’ADN et de conversion de bibliothèques ne permettent pas de récupérer toutes les molécules d’ADN potentiellement préservées. Par exemple, Kjaer et al. (11) ont découvert que l'ADN s'adsorbait préférentiellement sur les surfaces minérales argileuses par rapport aux surfaces non argileuses, en particulier sur la smectite, un minéral argileux, qui peut lier 200 fois plus d'ADN que le quartz et qui est un minéral courant dans les échantillons terrestres. Leur protocole d’extraction le plus performant a permis de récupérer 40 % de l’ADN lié au quartz et seulement 5 % de l’ADN lié à la smectite, ce qui suggère que la majeure partie de l’ADN était inaccessible. Bien qu’anecdotique, cette observation laisse entrevoir plusieurs opportunités pour améliorer la recherche sur l’ADN ancien, notamment en utilisant les caractérisations minéralogiques pour identifier les sites les plus prometteurs pour la récupération de l’ADN sédimentaire des époques anciennes et en affinant les approches expérimentales pour récupérer l’ADN lié à toutes les surfaces minérales. En l’absence d’amélioration des méthodes pour libérer l’ADN lié, l’évaluation microscopique des échantillons sédimentaires améliorera l’efficacité de la récupération de l’ADN. Massilani et coll. (47), par exemple, ont montré que l'ADN conservé dans les sédiments des grottes est concentré dans des particules microscopiques, en particulier des fragments d'os et de matières fécales conservés dans le substrat.
Les protocoles de conversion de bibliothèque pourraient également être rendus plus efficaces. Les protocoles optimisés de conversion de bibliothèques utilisent la ligature et la polymérisation enzymatiques, mais les extraits d'ADN anciens contiennent des inhibiteurs ainsi que des molécules présentant des dommages non caractérisés à l'ADN. Bien que nous puissions convertir aussi peu que 100 picogrammes d'ADN en bibliothèques en utilisant la méthode de Santa Cruz (25), il a été démontré que la préparation de bibliothèques ne convertit généralement qu'environ 10 à 50 % de l'ADN extrait (21), ce qui suggère que la plupart des molécules récupérées sont perdues au cours cette étape expérimentale. Les améliorations dans la préparation des bibliothèques peuvent inclure la conception d'enzymes plus robustes pour combattre les inhibiteurs ou le développement de protocoles intégrant la réparation enzymatique lors de la conversion des bibliothèques. De plus, réduire le recours aux étapes de ligase et de polymérase grâce à des stratégies enzymatiques alternatives, à la chimie bio-orthogonale ou au séquençage natif de l’ADN peut offrir de nouvelles approches pour convertir des molécules d’ADN actuellement non séquençables.
De nombreuses espèces qui sont des cibles évidentes pour la recherche sur l'ADN des temps très anciens sont éteintes et certaines, comme les canidés Xenocyon et les membres de base des familles des éléphants et des chevaux, n'ont pas de parent vivant proche sur le plan évolutif pour lequel un génome de référence idéal peut être produit. Cela présente des défis pour l’authentification et l’identification de l’ADN ancien ainsi que pour l’assemblage du génome guidé par référence. Bien que la longueur moyenne des fragments des séquences d'ADN très ancien soit courte, il peut être possible de générer des assemblages de novo à partir d'extraits anciens en capitalisant sur des méthodes qui utilisent la capture de la conformation des chromosomes pour conserver les informations de proximité utiles pour relier de courtes lectures au sein d'un chromosome (48). . Les approches consistant à séquencer l’ADN in situ (49) sont également prometteuses mais en sont actuellement aux premiers stades de développement. Les améliorations du traitement bioinformatique bénéficieront également à la reconstruction paléogénomique des eucaryotes et à l’identification de variants. Récemment, des génomes microbiens ont été assemblés à partir d'ADN récupéré à partir d'échantillons paléofécaux relativement récents (50) et de calculs dentaires archéologiques aussi vieux que 100 ka (51), suggérant une voie bioinformatique vers l'assemblage de novo de certains petits paléogénomes. Bien que cette approche ne s’applique probablement pas aux génomes des eucaryotes complexes, d’autres approches bioinformatiques peuvent améliorer la précision de ces assemblages à partir de données des séquences courtes. Le remplacement des génomes de référence linéaires mono-espèces par des graphiques de variation multi-espèces intégrant des variantes de plusieurs génomes (52), par exemple, peut augmenter le nombre de lectures se cartographiant sur un génome de référence. Cette approche présente l'avantage supplémentaire de permettre une variation entre les longueurs d’indels ( = insertion ou déletion ) ainsi qu'entre les nucléotides. Les approches d'assemblage itératif, telles que l'assembleur itératif de cartographie utilisé pour générer le premier génome mitochondrial de Néandertal (53), peuvent améliorer la cartographie vers des génomes plus complexes. Enfin, comme l'attribution taxonomique basée sur des références est toujours limitée aux séquences déposées dans des bases de données publiques, la population continue de ces bases de données continuera d'améliorer l'identification robuste de l'ADN récupéré des restes et des sédiments du Pléistocène inférieur et moyen.
Un défi considérable pour les études sur l’ADN des temps très anciens est de connaître l’âge des échantillons afin de pouvoir les placer dans des contextes évolutifs et géologiques plus larges. Comme l’ADN le plus ancien à ce jour provient d’organismes qui ont vécu au cours des dernières dizaines de milliers d’années, il est généralement possible d’estimer leur âge directement à l’aide de la datation au radiocarbone. Cependant, la courte demi-vie radioactive du carbone 14 (environ 6000 ans, ce qui est court à l’échelle géologique, car après 50000 ans, près de 10 demi-vies se sont écoulées, et il ne reste donc plus que 2-10 x la quantité de radioactivité initiale, c’est à dire environ 1 millième) signifie que les estimations d’âge sont souvent peu fiables si les organismes ont vécu plus de 50 ka. Les méthodes de datation à charge piégée, telles que la résonance de spin électronique (ESR) pour l'émail des dents ou les approches par luminescence pour les minéraux tels que le quartz et le feldspath, peuvent fournir des estimations d'âge pour des échantillons datant de tout le Pléistocène, mais exigent que les sédiments restent intacts depuis l'enfouissement [pour une revue, voir (54)]. Lorsque les protéines sont préservées, l’étendue de la racémisation, de l’hydrolyse et de la dégradation des acides aminés peut également estimer le temps écoulé depuis la mort, bien que l’on sache que les « horloges » des acides aminés varient selon les espèces et les localités (54).
Dans certains cas, des marqueurs paléoenvironnementaux, géologiques et géophysiques peuvent fournir des indices sur l'âge d'un échantillon. Un fossile pourrait être trouvé dans l'Arctique avec d'autres indicateurs paléoécologiques suggérant par exemple un environnement chaud et humide, indiquant que l'animal a vécu lors d'un précédent interglaciaire ou dans des sédiments à polarité inversée, ce qui suggère qu'il a vécu avant le dernier renversement paléomagnétique vers 780 ka. Dans certains environnements, les lits de téphra (couches de cendres volcaniques fines et déposées) peuvent être datés par des méthodes telles que la trace de fission du verre ou la datation argon-argon. Les lits de téphra, qui sont détectables même lorsqu'ils ne sont présents qu'en quantités microscopiques (55), ont joué un rôle particulièrement important dans la datation des carottes de sédiments, mais peuvent également fournir des indices contextuels sur l'âge des échantillons trouvés in situ sur les sites où le téphra est présent. Comme les éruptions volcaniques étaient courantes tout au long du Pléistocène, des téphrochronologies améliorées couvrant le Pléistocène inférieur et moyen aideront à placer l'ADN des temps anciens dans un contexte chronologique.
D’autres approches pour dater les génomes très anciens pourraient s’appuyer sur la nature prévisible du changement évolutif des organismes. Les méthodes d’horloge moléculaire déduisent l’âge des paléogénomes en estimant le degré d’évolution « manquante » le long d’une branche phylogénétique menant au paléogénome, souvent appelé « raccourcissement de branche » (56). L’accumulation de mutations étant approximativement constante dans le temps, les différences entre ces longueurs de branches devraient correspondre au nombre de générations qui séparent le paléogénome représenté des individus existants ou plus récents. Cependant, pour traduire les générations manquantes en temps calendaire, l’approche de raccourcissement des branches nécessite soit un calibrage indépendant des fossiles, soit une estimation de la durée d’une génération. Pour de nombreuses lignées ayant vécu au Pléistocène inférieur et moyen, les fossiles ancestraux datés sont rares et, en l’absence de parents proches vivants, les estimations de la durée d’une génération seraient imprécises. La variation des taux d’évolution entre lignées éloignées peut également réduire la puissance d’une approche comparative de datation moléculaire. Néanmoins, le développement d’approches utilisant les informations génomiques pour estimer l’âge des paléogénomes et leurs relations évolutives avec d’autres espèces constitue un domaine riche pour les recherches futures.
Conclusions
La prochaine décennie sera marquée par des progrès techniques continus qui élargiront la gamme taxonomique et géographique des paléogénomes des temps très lointains et des ensembles de données d’ADN sédimentaire de ces âges. Celles-ci incluront de nouvelles connaissances sur les substrats susceptibles de préserver l’ADN très âgé, ainsi que des approches raffinées pour libérer l’ADN lié à des matrices biologiques ou minéralogiques. Ces paléogénomes des temps reculés nouvellement assemblés seront placés dans un contexte chronologique grâce aux développements en géochronologie et en paléoécologie, ainsi qu'à des approches informatiques plus puissantes pour estimer l'âge des échantillons à l'aide d'une horloge moléculaire. Les ensembles de données des temps très anciens qui en résulteront permettront de reconstruire les histoires évolutives à travers des perturbations environnementales répétées, affinant ainsi la compréhension de l'évolution adaptative, de l'organisation communautaire et de la résilience des écosystèmes. De plus, comme le passé est par nature différent de tout ce qui existe aujourd’hui, l’accès à l’ADN des temps anciens offre de nombreuses opportunités de découverte scientifique, bien qu’encore imprévisibles.
Références
- J. Ehlers, P. L. Gibbard, P. D. Hughes, Quaternary Glaciations - Extent and Chronology: A Closer Look (Elsevier, 2011).
- C. Posth, G. Renaud, A. Mittnik, D. G. Drucker, H. Rougier, C. Cupillard, F. Valentin, C. Thevenet, A. Furtwängler, C. Wißing, M. Francken, M. Malina, M. Bolus, M. Lari, E. Gigli, G. Capecchi, I. Crevecoeur, C. Beauval, D. Flas, M. Germonpré, J. van der Plicht, R. Cottiaux, B. Gély, A. Ronchitelli, K. Wehrberger, D. Grigorescu, J. Svoboda, P. Semal, D. Caramelli, H. Bocherens, K. Harvati, N. J. Conard, W. Haak, A. Powell, J. Krause, Pleistocene Mitochondrial Genomes Suggest a Single Major Dispersal of Non-Africans and a Late Glacial Population Turnover in Europe. Curr. Biol. 26, 827–833 (2016).
- M. Baca, D. Popović, A. K. Agadzhanyan, K. Baca, N. J. Conard, H. Fewlass, T. Filek, M. Golubiński, I. Horáček, M. V. Knul, M. Krajcarz, M. Krokhaleva, L. Lebreton, A. Lemanik, L. C. Maul, D. Nagel, P. Noiret, J. Primault, L. Rekovets, S. E. Rhodes, A. Royer, N. V. Serdyuk, M. Soressi, J. R. Stewart, T. Strukova, S. Talamo, J. Wilczyński, A. Nadachowski, Ancient DNA of narrow-headed vole reveal common features of the Late Pleistocene population dynamics in cold-adapted small mammals. Proc. Biol. Sci. 290, 20222238 (2023).
- L. Loog, O. Thalmann, M. S. Sinding, V. J. Schuenemann, A. Perri, M. Germonpré, H. Bocherens, K. E. Witt, J. A. Samaniego Castruita, M. S. Velasco, I. K. C. Lundstrøm, N. Wales, G. Sonet, L. Frantz, H. Schroeder, J. Budd, E.-L. Jimenez, S. Fedorov, B. Gasparyan, A. W. Kandel, M. Lázničková-Galetová, H. Napierala, H.-P. Uerpmann, P. A. Nikolskiy, E. Y. Pavlova, V. V. Pitulko, K.-H. Herzig, R. S. Malhi, E. Willerslev, A. J. Hansen, K. Dobney, M. T. P. Gilbert, J. Krause, G. Larson, A. Eriksson, A. Manica, Ancient DNA suggests modern wolves trace their origin to a Late Pleistocene expansion from Beringia. Mol. Ecol. 29, 1596–1610 (2020).
- R. E. Green, J. Krause, A. W. Briggs, T. Maricic, U. Stenzel, M. Kircher, N. Patterson, H. Li, W. Zhai, M. H.-Y. Fritz, N. F. Hansen, E. Y. Durand, A.-S. Malaspinas, J. D. Jensen, T. Marques-Bonet, C. Alkan, K. Prüfer, M. Meyer, H. A. Burbano, J. M. Good, R. Schultz, A. Aximu-Petri, A. Butthof, B. Höber, B. Höffner, M. Siegemund, A. Weihmann, C. Nusbaum, E. S. Lander, C. Russ, N. Novod, J. Affourtit, M. Egholm, C. Verna, P. Rudan, D. Brajkovic, Ž. Kucan, I. Gušic, V. B. Doronichev, L. V. Golovanova, C. Lalueza-Fox, M. de la Rasilla, J. Fortea, A. Rosas, R. W. Schmitz, P. L. F. Johnson, E. E. Eichler, D. Falush, E. Birney, J. C. Mullikin, M. Slatkin, R. Nielsen, J. Kelso, M. Lachmann, D. Reich, S. Pääbo, A draft sequence of the Neandertal genome. Science 328, 710–722 (2010).
- A. D. Foote, K. Kaschner, S. E. Schultze, C. Garilao, S. Y. W. Ho, K. Post, T. F. G. Higham, C. Stokowska, H. van der Es, C. B. Embling, K. Gregersen, F. Johansson, E. Willerslev, M. T. P. Gilbert, Ancient DNA reveals that bowhead whale lineages survived Late Pleistocene climate change and habitat shifts. Nat. Commun. 4, 1677 (2013).
- C. Valdiosera, N. García, L. Dalén, C. Smith, R.-D. Kahlke, K. Lidén, A. Angerbjörn, J. L. Arsuaga, A. Götherström, Typing single polymorphic nucleotides in mitochondrial DNA as a way to access Middle Pleistocene DNA. Biol. Lett. 2, 601–603 (2006).
- L. Orlando, A. Ginolhac, G. Zhang, D. Froese, A. Albrechtsen, M. Stiller, M. Schubert, E. Cappellini, B. Petersen, I. Moltke, P. L. F. Johnson, M. Fumagalli, J. T. Vilstrup, M. Raghavan, T. Korneliussen, A.-S. Malaspinas, J. Vogt, D. Szklarczyk, C. D. Kelstrup, J. Vinther, A. Dolocan, J. Stenderup, A. M. V. Velazquez, J. Cahill, M. Rasmussen, X. Wang, J. Min, G. D. Zazula, A. Seguin-Orlando, C. Mortensen, K. Magnussen, J. F. Thompson, J. Weinstock, K. Gregersen, K. H. Røed, V. Eisenmann, C. J. Rubin, D. C. Miller, D. F. Antczak, M. F. Bertelsen, S. Brunak, K. A. S. Al-Rasheid, O. Ryder, L. Andersson, J. Mundy, A. Krogh, M. T. P. Gilbert, K. Kjær, T. Sicheritz-Ponten, L. J. Jensen, J. V. Olsen, M. Hofreiter, R. Nielsen, B. Shapiro, J. Wang, E. Willerslev, Recalibrating Equus evolution using the genome sequence of an early Middle Pleistocene horse. Nature 499, 74–78 (2013).
- A. Barlow, J. L. A. Paijmans, F. Alberti, B. Gasparyan, G. Bar-Oz, R. Pinhasi, I. Foronova, A. Y. Puzachenko, M. Pacher, L. Dalén, G. Baryshnikov, M. Hofreiter, Middle Pleistocene genome calibrates a revised evolutionary history of extinct cave bears. Curr. Biol. 31, 1771–1779 (2021).
- T. van der Valk, P. Pečnerová, D. Díez-Del-Molino, A. Bergström, J. Oppenheimer, S. Hartmann, G. Xenikoudakis, J. A. Thomas, M. Dehasque, E. Sağlıcan, F. R. Fidan, I. Barnes, S. Liu, M. Somel, P. D. Heintzman, P. Nikolskiy, B. Shapiro, P. Skoglund, M. Hofreiter, A. M. Lister, A. Götherström, L. Dalén, Million-year-old DNA sheds light on the genomic history of mammoths. Nature 591, 265–269 (2021).
- K. H. Kjær, M. Winther Pedersen, B. De Sanctis, B. De Cahsan, T. S. Korneliussen, C. S. Michelsen, K. K. Sand, S. Jelavić, A. H. Ruter, A. M. A. Schmidt, K. K. Kjeldsen, A. S. Tesakov, I. Snowball, J. C. Gosse, I. G. Alsos, Y. Wang, C. Dockter, M. Rasmussen, M. E. Jørgensen, B. Skadhauge, A. Prohaska, J. Å. Kristensen, M. Bjerager, M. E. Allentoft, E. Coissac, A. Rouillard, A. Simakova, A. Fernandez-Guerra, C. Bowler, M. Macias-Fauria, L. Vinner, J. J. Welch, A. J. Hidy, M. Sikora, M. J. Collins, R. Durbin, N. K. Larsen, E. Willerslev, PhyloNorway Consortium, A 2-million-year-old ecosystem in Greenland uncovered by environmental DNA. Nature 612, 283–291 (2022).
- J. Courtin, A. Perfumo, A. A. Andreev, T. Opel, K. R. Stoof-Leichsenring, M. E. Edwards, J. B. Murton, U. Herzschuh, Pleistocene glacial and interglacial ecosystems inferred from ancient DNA analyses of permafrost sediments from Batagay megaslump, East Siberia. Environ. DNA 4, 1265–1283 (2022).
- E. Willerslev, A. J. Hansen, J. Binladen, T. B. Brand, M. T. P. Gilbert, B. Shapiro, M. Bunce, C. Wiuf, D. A. Gilichinsky, A. Cooper, Diverse plant and animal genetic records from Holocene and Pleistocene sediments. Science 300, 791–795 (2003).
- E. Willerslev, E. Cappellini, W. Boomsma, R. Nielsen, M. B. Hebsgaard, T. B. Brand, M. Hofreiter, M. Bunce, H. N. Poinar, D. Dahl-Jensen, S. Johnsen, J. P. Steffensen, O. Bennike, J.-L. Schwenninger, R. Nathan, S. Armitage, C.-J. de Hoog, V. Alfimov, M. Christl, J. Beer, R. Muscheler, J. Barker, M. Sharp, K. E. H. Penkman, J. Haile, P. Taberlet, M. T. P. Gilbert, A. Casoli, E. Campani, M. J. Collins, Ancient biomolecules from deep ice cores reveal a forested southern Greenland. Science 317, 111–114 (2007).
- M. Meyer, Q. Fu, A. Aximu-Petri, I. Glocke, B. Nickel, J.-L. Arsuaga, I. Martínez, A. Gracia, J. M. B. de Castro, E. Carbonell, S. Pääbo, A mitochondrial genome sequence of a hominin from Sima de los Huesos. Nature 505, 403–406 (2014).
- E. I. Zavala, Z. Jacobs, B. Vernot, M. V. Shunkov, M. B. Kozlikin, A. P. Derevianko, E. Essel, C. de Fillipo, S. Nagel, J. Richter, F. Romagné, A. Schmidt, B. Li, K. O’Gorman, V. Slon, J. Kelso, S. Pääbo, R. G. Roberts, M. Meyer, Pleistocene sediment DNA reveals hominin and faunal turnovers at Denisova Cave. Nature 595, 399–403 (2021).
- T. Lindahl, Instability and decay of the primary structure of DNA. Nature 362, 709–715 (1993).
- J. S. Pedersen, E. Valen, A. M. V. Velazquez, B. J. Parker, M. Rasmussen, S. Lindgreen, B. Lilje, D. J. Tobin, T. K. Kelly, S. Vang, R. Andersson, P. A. Jones, C. A. Hoover, A. Tikhonov, E. Prokhortchouk, E. M. Rubin, A. Sandelin, M. T. P. Gilbert, A. Krogh, E. Willerslev, L. Orlando, Genome-wide nucleosome map and cytosine methylation levels of an ancient human genome. Genome Res. 24, 454–466 (2014).
- A. J. Hansen, D. L. Mitchell, C. Wiuf, L. Paniker, T. B. Brand, J. Binladen, D. A. Gilichinsky, R. Rønn, E. Willerslev, Crosslinks rather than strand breaks determine access to ancient DNA sequences from frozen sediments. Genetics 173, 1175–1179 (2006).
- A. W. Briggs, U. Stenzel, P. L. F. Johnson, R. E. Green, J. Kelso, K. Prüfer, M. Meyer, J. Krause, M. T. Ronan, M. Lachmann, S. Pääbo, Patterns of damage in genomic DNA sequences from a Neandertal. Proc. Natl. Acad. Sci. U.S.A. 104, 14616–14621 (2007).
- L. Bokelmann, I. Glocke, M. Meyer, Reconstructing double-stranded DNA fragments on a single-molecule level reveals patterns of degradation in ancient samples. Genome Res. 30, 1449–1457 (2020).
- J. Dabney, M. Meyer, S. Pääbo, Ancient DNA damage. Cold Spring Harb. Perspect. Biol. 5, a012567 (2013).
- E. Hempel, F. Bibi, J. T. Faith, K.-P. Koepfli, A. M. Klittich, D. A. Duchêne, J. S. Brink, D. C. Kalthoff, L. Dalén, M. Hofreiter, M. V. Westbury, Blue Turns to Gray: Paleogenomic Insights into the Evolutionary History and Extinction of the Blue Antelope (Hippotragus leucophaeus). Mol. Biol. Evol. 39, msac241 (2022).
- M.-T. Gansauge, T. Gerber, I. Glocke, P. Korlević, L. Lippik, S. Nagel, L. M. Riehl, A. Schmidt, M. Meyer, Single-stranded DNA library preparation from highly degraded DNA using T4 DNA ligase. Nucleic Acids Res. 45, e79–e79 (2017).
- J. D. Kapp, R. E. Green, B. Shapiro, A Fast and Efficient Single-stranded Genomic Library Preparation Method Optimized for Ancient DNA. J. Hered. 112, 241–249 (2021).
- A. W. Briggs, U. Stenzel, M. Meyer, J. Krause, M. Kircher, S. Pääbo, Removal of deaminated cytosines and detection of in vivo methylation in ancient DNA. Nucleic Acids Res. 38, e87 (2010).
- C. de Filippo, M. Meyer, K. Prüfer, Quantifying and reducing spurious alignments for the analysis of ultra-short ancient DNA sequences. BMC Biol. 16, 121 (2018).
- K. Prüfer, snpAD: An ancient DNA genotype caller. Bioinformatics 34, 4165–4171 (2018).
- S. Peyrégne, V. Slon, F. Mafessoni, C. de Filippo, M. Hajdinjak, S. Nagel, B. Nickel, E. Essel, A. Le Cabec, K. Wehrberger, N. J. Conard, C. J. Kind, C. Posth, J. Krause, G. Abrams, D. Bonjean, K. Di Modica, M. Toussaint, J. Kelso, M. Meyer, S. Pääbo, K. Prüfer, Nuclear DNA from two early Neandertals reveals 80,000 years of genetic continuity in Europe. Sci. Adv. 5, eaaw5873 (2019).
- W. Miller, S. C. Schuster, A. J. Welch, A. Ratan, O. C. Bedoya-Reina, F. Zhao, H. L. Kim, R. C. Burhans, D. I. Drautz, N. E. Wittekindt, L. P. Tomsho, E. Ibarra-Laclette, L. Herrera-Estrella, E. Peacock, S. Farley, G. K. Sage, K. Rode, M. Obbard, R. Montiel, L. Bachmann, O. Ingólfsson, J. Aars, T. Mailund, O. Wiig, S. L. Talbot, C. Lindqvist, Polar and brown bear genomes reveal ancient admixture and demographic footprints of past climate change. Proc. Natl. Acad. Sci. U.S.A. 109, E2382–E2390 (2012).
- J. A. Cahill, P. D. Heintzman, K. Harris, M. D. Teasdale, J. Kapp, A. E. R. Soares, I. Stirling, D. Bradley, C. J. Edwards, K. Graim, A. A. Kisleika, A. V. Malev, N. Monaghan, R. E. Green, B. Shapiro, Genomic Evidence of Widespread Admixture from Polar Bears into Brown Bears during the Last Ice Age. Mol. Biol. Evol. 35, 1120–1129 (2018).
- M.-S. Wang, G. G. R. Murray, D. Mann, P. Groves, A. O. Vershinina, M. A. Supple, J. D. Kapp, R. Corbett-Detig, S. E. Crump, I. Stirling, K. L. Laidre, M. Kunz, L. Dalén, R. E. Green, B. Shapiro, A polar bear paleogenome reveals extensive ancient gene flow from polar bears into brown bears. Nat. Ecol. Evol. 6, 936–944 (2022).
- J. T. Weir, D. Schluter, Ice sheets promote speciation in boreal birds. Proc. Biol. Sci. 271, 1881–1887 (2004).
- A. M. Lister, The impact of Quaternary Ice Ages on mammalian evolution. Philos. Trans. R. Soc. B, 221–241 (2004).
- M. Meyer, J.-L. Arsuaga, C. de Filippo, S. Nagel, A. Aximu-Petri, B. Nickel, I. Martínez, A. Gracia, J. M. Bermúdez de Castro, E. Carbonell, B. Viola, J. Kelso, K. Prüfer, S. Pääbo, Nuclear DNA sequences from the Middle Pleistocene Sima de los Huesos hominins. Nature 531, 504–507 (2016).
- D. Froese, M. Stiller, P. D. Heintzman, A. V. Reyes, G. D. Zazula, A. E. R. Soares, M. Meyer, E. Hall, B. J. L. Jensen, L. J. Arnold, R. D. E. MacPhee, B. Shapiro, Fossil and genomic evidence constrains the timing of bison arrival in North America. Proc. Natl. Acad. Sci. U.S.A. 114, 3457–3462 (2017).
- D. Díez-Del-Molino, M. Dehasque, J. C. Chacón-Duque, P. Pečnerová, A. Tikhonov, A. Protopopov, V. Plotnikov, F. Kanellidou, P. Nikolskiy, P. Mortensen, G. K. Danilov, S. Vartanyan, M. T. P. Gilbert, A. M. Lister, P. D. Heintzman, T. van der Valk, L. Dalén, Genomics of adaptive evolution in the woolly mammoth. Curr. Biol. 33, 1753–1764.e4 (2023).
- P. U. Clark, D. Archer, D. Pollard, J. D. Blum, J. A. Rial, V. Brovkin, A. C. Mix, N. G. Pisias, M. Roy, The middle Pleistocene transition: Characteristics, mechanisms, and implications for long-term changes in atmospheric pCO2. Quat. Sci. Rev. 25, 3150–3184 (2006).
- J. R. Stewart, A. M. Lister, I. Barnes, L. Dalén, Refugia revisited: Individualistic responses of species in space and time. Proc. Biol. Sci. 277, 661–671 (2010).
- E. Lord, A. Marangoni, M. Baca, D. Popović, A. V. Goropashnaya, J. R. Stewart, M. V. Knul, P. Noiret, M. Germonpré, E.-L. Jimenez, N. I. Abramson, S. Vartanyan, S. Prost, N. G. Smirnov, E. A. Kuzmina, R.-A. Olsen, V. B. Fedorov, L. Dalén, Population dynamics and demographic history of Eurasian collared lemmings. BMC Ecol. Evol. 22, 126 (2022).
- J. Ortego, L. L. Knowles, Geographical isolation versus dispersal: Relictual alpine grasshoppers support a model of interglacial diversification with limited hybridization. Mol. Ecol. 31, 296–312 (2022).
- L. E. Lisiecki, M. E. Raymo, L. E. Lisiecki, M. E. Raymo, A Pliocene-Pleistocene stack of 57 globally distributed benthic δ18O records. Paleoceanography 20, (2005).
- L. Armbrecht, M. E. Weber, M. E. Raymo, V. L. Peck, T. Williams, J. Warnock, Y. Kato, I. Hernández-Almeida, F. Hoem, B. Reilly, S. Hemming, I. Bailey, Y. M. Martos, M. Gutjahr, V. Percuoco, C. Allen, S. Brachfeld, F. G. Cardillo, Z. Du, G. Fauth, C. Fogwill, M. Garcia, A. Glüder, M. Guitard, J.-H. Hwang, M. Iizuka, B. Kenlee, S. O’Connell, L. F. Pérez, T. A. Ronge, O. Seki, L. Tauxe, S. Tripathi, X. Zheng, Ancient marine sediment DNA reveals diatom transition in Antarctica. Nat. Commun. 13, 5787 (2022).
- A. J. Monteath, S. Kuzmina, M. Mahony, F. Calmels, T. Porter, R. Mathewes, P. Sanborn, G. Zazula, B. Shapiro, T. J. Murchie, H. N. Poinar, T. Sadoway, E. Hall, S. Hewitson, D. Froese, Relict permafrost preserves megafauna, insects, pollen, soils and pore-ice isotopes of the mammoth steppe and its collapse in central Yukon. Quat. Sci. Rev. 299, 107878 (2023).
- C. L. Clarke, M. E. Edwards, L. Gielly, D. Ehrich, P. D. M. Hughes, L. M. Morozova, H. Haflidason, J. Mangerud, J. I. Svendsen, I. G. Alsos, Persistence of arctic-alpine flora during 24,000 years of environmental change in the Polar Urals. Sci. Rep. 9, 19613 (2019).
- M. W. Pedersen, B. De Sanctis, N. F. Saremi, M. Sikora, E. E. Puckett, Z. Gu, K. L. Moon, J. D. Kapp, L. Vinner, Z. Vardanyan, C. F. Ardelean, J. Arroyo-Cabrales, J. A. Cahill, P. D. Heintzman, G. Zazula, R. D. E. MacPhee, B. Shapiro, R. Durbin, E. Willerslev, Environmental genomics of Late Pleistocene black bears and giant short-faced bears. Curr. Biol. 31, 2728–2736.e8 (2021).
- D. Massilani, M. W. Morley, S. M. Mentzer, V. Aldeias, B. Vernot, C. Miller, M. Stahlschmidt, M. B. Kozlikin, M. V. Shunkov, A. P. Derevianko, N. J. Conard, S. Wurz, C. S. Henshilwood, J. Vasquez, E. Essel, S. Nagel, J. Richter, B. Nickel, R. G. Roberts, S. Pääbo, V. Slon, P. Goldberg, M. Meyer, Microstratigraphic preservation of ancient faunal and hominin DNA in Pleistocene cave sediments. Proc. Natl. Acad. Sci. U.S.A. 119, e2113666118 (2022).
- N. Kaplan, J. Dekker, High-throughput genome scaffolding from in vivo DNA interaction frequency. Nat. Biotechnol. 31, 1143–1147 (2013).
- A. C. Payne, Z. D. Chiang, P. L. Reginato, S. M. Mangiameli, E. M. Murray, C.-C. Yao, S. Markoulaki, A. S. Earl, A. S. Labade, R. Jaenisch, G. M. Church, E. S. Boyden, J. D. Buenrostro, F. Chen, In situ genome sequencing resolves DNA sequence and structure in intact biological samples. Science 371, eaay3446 (2021).
- M. C. Wibowo, Z. Yang, M. Borry, A. Hübner, K. D. Huang, B. T. Tierney, S. Zimmerman, F. Barajas-Olmos, C. Contreras-Cubas, H. García-Ortiz, A. Martínez-Hernández, J. M. Luber, P. Kirstahler, T. Blohm, F. E. Smiley, R. Arnold, S. A. Ballal, S. J. Pamp, J. Russ, F. Maixner, O. Rota-Stabelli, N. Segata, K. Reinhard, L. Orozco, C. Warinner, M. Snow, S. LeBlanc, A. D. Kostic, Reconstruction of ancient microbial genomes from the human gut. Nature 594, 234–239 (2021).
- M. Klapper, A. Hübner, A. Ibrahim, I. Wasmuth, M. Borry, V. G. Haensch, S. Zhang, W. K. Al-Jammal, H. Suma, J. A. Fellows Yates, J. Frangenberg, I. M. Velsko, S. Chowdhury, R. Herbst, E. V. Bratovanov, H.-M. Dahse, T. Horch, C. Hertweck, M. R. González Morales, L. G. Straus, I. Vilotijevic, C. Warinner, P. Stallforth, Natural products from reconstructed bacterial genomes of the Middle and Upper Paleolithic. Science 380, 619–624 (2023).
- R. Martiniano, E. Garrison, E. R. Jones, A. Manica, R. Durbin, Removing reference bias and improving indel calling in ancient DNA data analysis by mapping to a sequence variation graph. Genome Biol. 21, 250 (2020).
- R. E. Green, A.-S. Malaspinas, J. Krause, A. W. Briggs, P. L. F. Johnson, C. Uhler, M. Meyer, J. M. Good, T. Maricic, U. Stenzel, K. Prüfer, M. Siebauer, H. A. Burbano, M. Ronan, J. M. Rothberg, M. Egholm, P. Rudan, D. Brajković, Z. Kućan, I. Gusić, M. Wikström, L. Laakkonen, J. Kelso, M. Slatkin, S. Pääbo, A complete Neandertal mitochondrial genome sequence determined by high-throughput sequencing. Cell 134, 416–426 (2008).
- K. E. H. Penkman, G. A. T. Duller, H. M. Roberts, D. Colarossi, M. R. Dickinson, D. White, Dating the Paleolithic: Trapped charge methods and amino acid geochronology. Proc. Natl. Acad. Sci. U.S.A. 119, e2109324119 (2022).
- S. M. Davies, Cryptotephras: The revolution in correlation and precision dating. J. Quat. Sci. 30, 114–130 (2015).
- M. Meyer, M. Kircher, M.-T. Gansauge, H. Li, F. Racimo, S. Mallick, J. G. Schraiber, F. Jay, K. Prüfer, C. de Filippo, P. H. Sudmant, C. Alkan, Q. Fu, R. Do, N. Rohland, A. Tandon, M. Siebauer, R. E. Green, K. Bryc, A. W. Briggs, U. Stenzel, J. Dabney, J. Shendure, J. Kitzman, M. F. Hammer, M. V. Shunkov, A. P. Derevianko, N. Patterson, A. M. Andrés, E. E. Eichler, M. Slatkin, D. Reich, J. Kelso, S. Pääbo, A high-coverage genome sequence from an archaic Denisovan individual. Science 338, 222–226 (2012).
- J. A. Fellows Yates, A. Andrades Valtueña, Å. J. Vågene, B. Cribdon, I. M. Velsko, M. Borry, M. J. Bravo-Lopez, A. Fernandez-Guerra, E. J. Green, S. L. Ramachandran, P. D. Heintzman, M. A. Spyrou, A. Hübner, A. S. Gancz, J. Hider, A. F. Allshouse, V. Zaro, C. Warinner, Community-curated and standardised metadata of published ancient metagenomic samples with AncientMetagenomeDir. Sci. Data 8, 31 (2021).
- J. Von Eggers, M.-E. Monchamp, E. Capo, C. Giguet-Covex, T. Spanbauer, P. D. Heintzman, Inventory of ancient environmental DNA from sedimentary archives: locations, methods, and target taxa, Version 1.0, Zenodo (2022);
https://zenodo.org/record/6847522. - D. Diez-del-Molino, P. D. Heintzman, L. Dalén, A list of representative paleogenomic datasets derived from human and faunal remains, Version 1.0, Zenodo (2023);
https://zenodo.org/record/8270285. - J. Dabney, M. Knapp, I. Glocke, M.-T. Gansauge, A. Weihmann, B. Nickel, C. Valdiosera, N. García, S. Pääbo, J.-L. Arsuaga, M. Meyer, Complete mitochondrial genome sequence of a Middle Pleistocene cave bear reconstructed from ultrashort DNA fragments. Proc. Natl. Acad. Sci. U.S.A. 110, 15758–15763 (2013).
- A. M. Lister, On the type material and evolution of North American mammoths. Quat. Int. 443, 14–31 (2017).