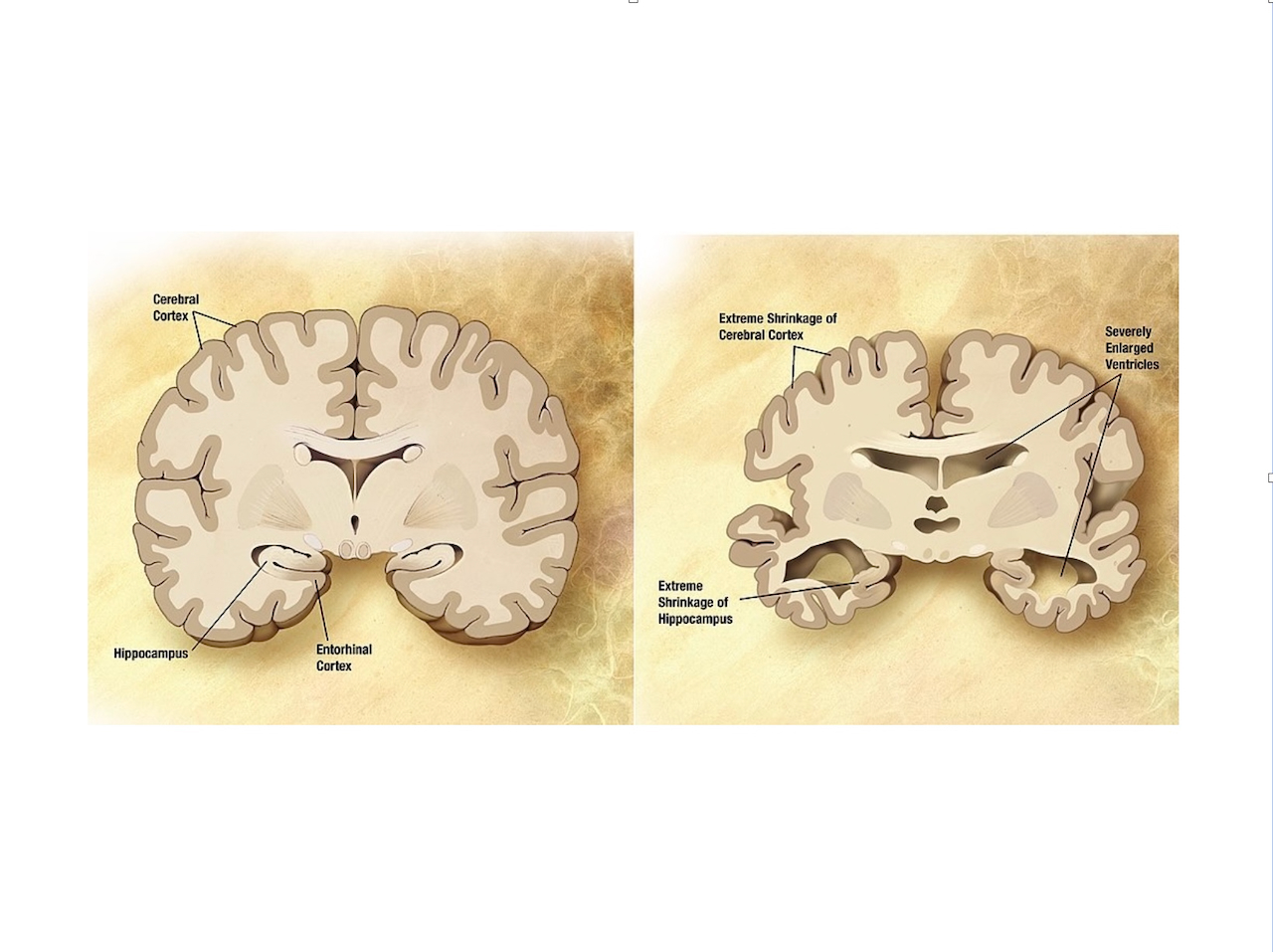
SCIENCE - LE FARDEAU D’UN GÈNE - 12 SEPTEMBRE 2024
Une variante appelée APOE4 est connue pour son lien avec la maladie d’Alzheimer.
De nouvelles connaissances sur sa fonction peuvent-elles aider à prévenir la maladie ?
PAR JOCELYN KAISER, journaliste scientifique

Ray Smith, 70 ans (à gauche), et Carol Turner, 68 ans (à droite), avec leur mère Mattie Smith, 90 ans, atteinte de la maladie d’Alzheimer.
Ray Smith a hérité de deux copies du gène de risque d’Alzheimer APOE4 ; Turner en possède une copie.
BILL TIERNAN
Carol Turner et son frère Ray Smith ont pu voir de près ce que c’est que de vivre avec la maladie d’Alzheimer. En 2020, leur père est décédé à l’âge de 93 ans dans un service de démence, après avoir passé des mois à voir sa famille à travers une fenêtre à cause de la pandémie de COVID-19. Peu de temps après, leur mère a montré des signes : elle avait du mal à dormir, elle ne reconnaissait pas certains visages et sa recette de brownie préférée ne fonctionnait plus tout à fait. Des scanners cérébraux ont révélé une abondance de plaques de bêta-amyloïde, la protéine collante caractéristique de la maladie d’Alzheimer.
Compte tenu de ces antécédents familiaux, le gériatre de leur mère à l’école de médecine de l’est de la Virginie, à proximité de Norfolk, savait que les frères et sœurs pouvaient être à haut risque de développer la maladie d’Alzheimer. Il leur a indiqué un essai clinique pour lequel un site de la faculté de médecine recherchait des participants pour tester un traitement préventif. Là, des tests ADN ont révélé des nouvelles plus inquiétantes. Smith, aujourd’hui âgé de 70 ans, est porteur de deux copies de la variante génétique la plus courante – et peut-être la plus fascinante sur le plan scientifique – des variantes plus étroitement liées à la maladie d’Alzheimer à un stade avancé de la vie : APOE4. Turner, 68 ans, en est porteur d’une copie.
Cette variante est devenue un sujet de préoccupation majeur depuis que la découverte qu’elle augmente considérablement le risque de maladie d’Alzheimer a électrisé le domaine il y a plus de 30 ans. Les 164 millions de personnes dans le monde qui sont porteuses de deux copies d’APOE4, l’une des trois variantes du gène APOE, peuvent avoir un risque de développer une maladie d’Alzheimer à un stade tardif de huit à 25 fois plus élevé que celles qui ne sont porteuses que de la version la plus courante du gène, APOE3. Environ 1,6 milliard de personnes ont une seule copie de cette variante, dont l’auteur de cet article. Pour nous, le risque de maladie peut également être multiplié par trois ou plus, selon l’ascendance de la personne.
La découverte en 1993 du rôle de l’APOE4 dans la maladie d’Alzheimer semblait prometteuse de nouvelles perspectives sur les racines de cette maladie déconcertante, et peut-être une nouvelle cible thérapeutique. Mais les progrès ont été lents. La protéine codée par l’APOE, l’apolipoprotéine E (ApoE), joue de nombreux rôles dans le cerveau, ce qui rend difficile de distinguer ceux qui sont pertinents pour le développement de la maladie et pourquoi certaines variantes de l’APOE augmentent le risque. Mais les scientifiques pourraient enfin gagner du terrain. De nouvelles connaissances sur les effets de l’APOE, ainsi qu’une vague d’études sur des versions protectrices rares, alimentent l’intérêt pour des thérapies potentielles qui contreraient les effets nocifs de l’APOE4.
Une étude publiée par des chercheurs espagnols en mai dans Nature Medicine, qui renforce l’idée que l’APOE4 devrait être considérée non seulement comme un facteur de risque de la maladie, mais comme une cause directe, rend ces efforts encore plus urgents. L’étude a révélé que sur les 2 % de personnes d’origine européenne possédant deux copies de l’APOE4, 75 % présenteront une accumulation d’amyloïde sur les scanners cérébraux (mais pas nécessairement de démence) à l’âge de 65 ans. Le rôle de cette variante dans la maladie est « devenu de plus en plus clair », explique Yadong Huang, expert de l’APOE4 aux Instituts Gladstone, qui a rédigé un commentaire d’accompagnement.
De telles découvertes ont stimulé le petit groupe de chercheurs sur la maladie d’Alzheimer qui ont consacré leur carrière à l’APOE4. « L’APOE joue un rôle important non seulement dans le risque d’Alzheimer, mais aussi probablement dans la progression de la maladie », déclare le neurologue et neuroscientifique David Holtzman de l’Université de Washington à Saint-Louis. « C’est vraiment fantastique que les gens cherchent maintenant des moyens de la cibler. »
Après avoir appris qu’elle et son frère étaient porteurs du « gène d’Alzheimer » et qu’ils souffraient déjà d’amyloïde cérébrale, Turner s’est inquiétée. « Le médecin a dit : "Vous et votre frère allez probablement l’attraper plus tard si vous ne faites rien". » Fin 2021, Turner est devenue la première Afro-Américaine à participer à l’essai, dans lequel les personnes présentant des niveaux importants d’amyloïde mais aucun symptôme d’Alzheimer reçoivent soit un placebo, soit des perfusions bihebdomadaires ou mensuelles d’un anticorps éliminant l’amyloïde, appelé lecanemab pour peut-être retarder la maladie. Smith a rejoint l’essai peu de temps après. Plus de 2 ans après le début du traitement de 4 ans, tous deux disent n’avoir remarqué aucun déclin de leur mémoire.
L’APOE ÉTAIT À L’ORIGINE connue pour son rôle dans la régulation du cholestérol sanguin, qui contribue aux maladies cardiovasculaires. (La plupart des personnes porteuses de la variante génétique APOE2 ont un faible taux de cholestérol et sont protégées contre les maladies cardiaques, alors que les porteurs de l’APOE4 sont les plus à risque.) Mais au début des années 1990, des chercheurs de l’Université Duke ont identifié une région de l’ADN liée à un risque plus élevé de maladie d’Alzheimer à apparition tardive, qui commence après 65 ans et est la forme la plus courante de la maladie. L’équipe de Duke et d’autres ont également découvert un autre indice : la protéine ApoE, dont le gène se trouvait dans la région suspecte, était présente dans les plaques amyloïdes soupçonnées de provoquer des lésions neurologiques dans la maladie d’Alzheimer.
En 1993, le groupe de la généticienne de Duke Margaret Pericak-Vance a identifié la variante à risque APOE4 en comparant les gènes des personnes qui ont développé une maladie d’Alzheimer à apparition tardive avec ceux de leurs proches non touchés, soit 234 personnes issues de 42 familles au total.
Cette découverte, publiée dans Science, a été annoncée comme une avancée majeure dans le domaine. Le neurologue Allen Roses, qui a dirigé l’équipe chargée d’étudier l’APOE à Duke, a prédit dans le New York Times que « dans 10 à 15 ans, nous disposerons d’un médicament sûr et efficace qu’une personne de 50 ans pourrait prendre tous les jours pour prévenir la maladie d’Alzheimer ».
Des études de plus grande envergure ont rapidement révélé que les porteurs d’APOE4 accumulaient plus d’amyloïde plus tôt dans la vie que les non-porteurs, bien avant de développer les symptômes de la maladie d’Alzheimer. Le groupe de Duke et d’autres ont fait la découverte tout aussi convaincante que les personnes ayant une ou deux copies d’une variante différente, APOE2, développaient peu d’amyloïde et étaient protégées de la maladie. Des différences intrigantes basées sur l’ascendance sont également apparues : les risques pour les Afro-Américains porteurs d’APOE4 sont légèrement inférieurs à ceux des personnes ayant des ancêtres européens, tandis que chez les Asiatiques de l’Est, le risque est beaucoup plus élevé, jusqu’à 25 fois plus élevé pour les personnes ayant deux copies, par rapport à celles ayant deux gènes APOE3.
Et pourtant, malgré près de 7 000 articles publiés sur APOE4 et la maladie d’Alzheimer, la prédiction de Roses concernant un traitement sûr et efficace ciblant la protéine reste un espoir lointain. Cela est en partie dû au fait que l’ApoE a de multiples sources dans le cerveau et joue divers rôles. La protéine est principalement fabriquée par les astrocytes, des cellules qui aident à nourrir et à entretenir les neurones, ainsi que par les cellules microgliales, les cellules immunitaires du cerveau. En cas de stress, les neurones en produisent également.
Des travaux commencés dans les années 1980 ont révélé que la principale fonction de l’ApoE dans le cerveau est d’aider au transport et au traitement des lipides, essentiels à la capacité des cellules à réparer les dommages, à transmettre des signaux, etc. La protéine se lie au cholestérol et à d’autres lipides pour former des particules qui sont délivrées aux cellules cérébrales, qui les décomposent ensuite en formes utilisables par les cellules.
Des études ont également montré que l’ApoE aide à éliminer l’amyloïde, une tâche que l’ApoE4, qui diffère de l’ApoE2 et de l’ApoE3 par seulement deux acides aminés, fait moins efficacement. Le cerveau des porteurs de l’APOE4 développe non seulement plus d’amyloïde que celui des non-porteurs, mais est également moins capable de briser les plaques si elles se forment. Ils sont également plus susceptibles de produire des enchevêtrements mal repliés d’une protéine appelée tau qui contribuerait à la maladie. Mais ce ne sont pas les seules voies possibles par lesquelles APOE4 pourrait provoquer la maladie d’Alzheimer : les chercheurs ont également découvert que la protéine de cette variante altère les centrales énergétiques cellulaires appelées mitochondries, rend la barrière hémato-encéphalique plus perméable aux toxines, entrave le transport et le métabolisme des lipides et accélère l’inflammation, ce qui endommage les neurones.
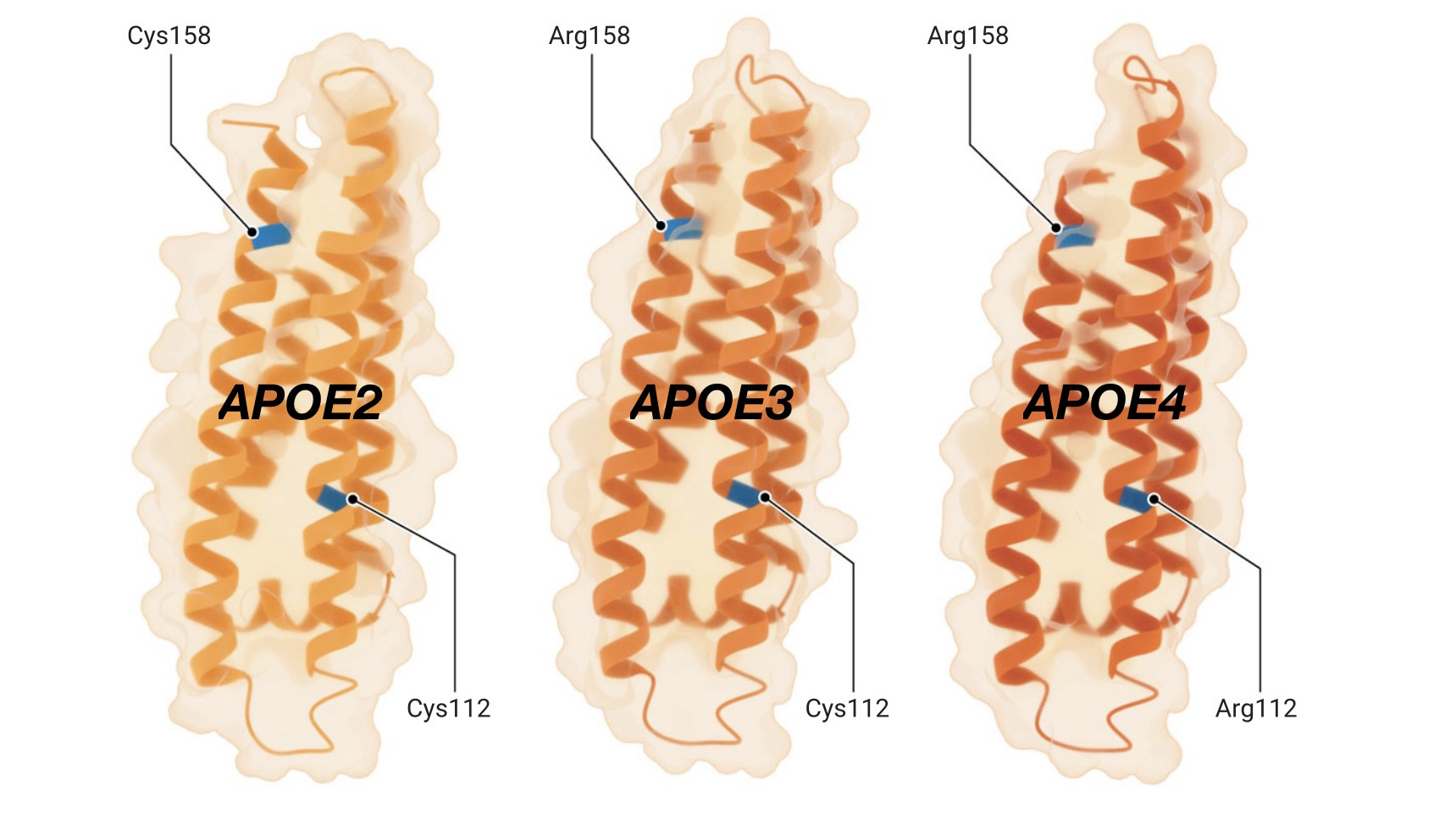
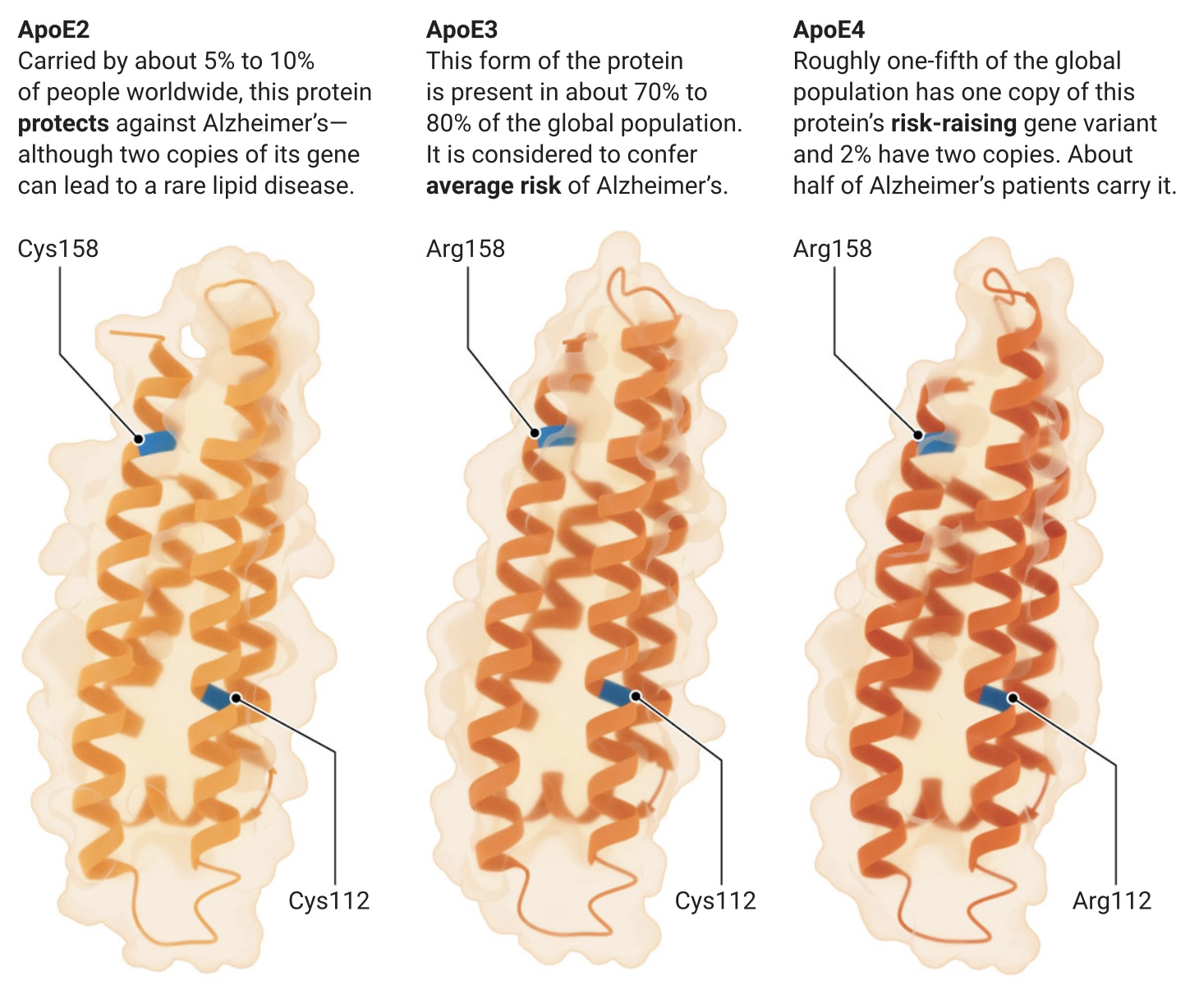
De petites différences, de grands effets
Les trois formes de protéines codées par les variantes génétiques APOE2, APOE3 et APOE4 ne diffèrent que d’un ou deux acides aminés (en bleu, ci-dessous), mais entraînent des différences considérables dans le risque de maladie d’Alzheimer.
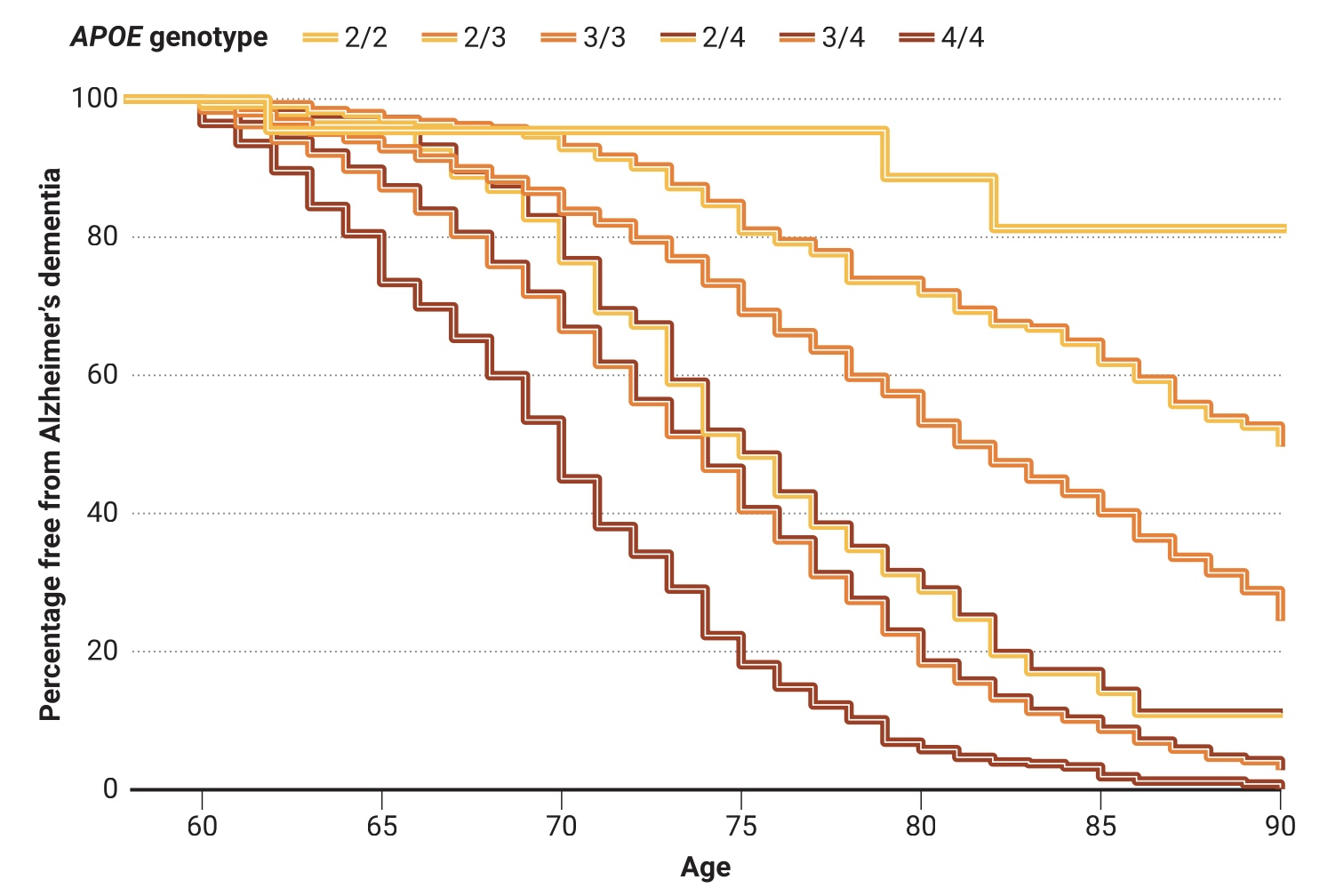
Le hasard du tirage au sort
Différentes combinaisons des trois variantes du gène APOE (APOE2, APOE3 et APOE4) augmentent ou diminuent le risque de maladie d’Alzheimer à un âge avancé. Ces courbes sont issues d’une étude de 2020 portant sur 5 000 personnes blanches non hispaniques décédées, atteintes ou non de la maladie d’Alzheimer. De telles études rétrospectives peuvent exagérer les risques ; dans les études qui ont suivi des personnes cognitivement normales au fil du temps, au moins 40 % des porteurs d’APOE4/4 n’étaient toujours pas atteints de démence à 85 ans.
Holtzman soupçonne que parmi ces nombreux effets, « il n’y en a probablement que quelques-uns qui ont vraiment un impact majeur sur la maladie ». Mais le défi d’identifier les effets pertinents et de démêler leurs mécanismes a dissuadé les développeurs de médicaments potentiels. Le fait que les chercheurs ne sachent pas exactement quel effet un traitement devrait avoir n’a pas aidé. Si l’ApoE4 causait des dommages parce qu’il était trop faible pour éliminer l’amyloïde, une thérapie idéale devrait augmenter ses niveaux dans le cerveau. En revanche, s’il était intrinsèquement nocif, un traitement devrait le freiner.
Des chercheurs affirment que l’intérêt pour l’APOE4 a également diminué parce que le domaine de la maladie d’Alzheimer était fixé sur l’amyloïde comme cause principale de la maladie. « Nous étions en concurrence avec l’hypothèse de l’amyloïde », en particulier l’idée selon laquelle l’élimination directe des plaques pourrait traiter la maladie, explique Pericak-Vance, aujourd’hui à l’Université de Miami. Des milliards de dollars de recherche publique et privée ont été consacrés à tenter de montrer que les anticorps anti-amyloïdes pouvaient ralentir la maladie d’Alzheimer.
Ces travaux ont conduit à l’approbation aux États-Unis du lécanemab en 2023, et d’un deuxième médicament, le donanemab, en juillet, pour les personnes atteintes de la maladie d’Alzheimer au stade précoce. Mais les avantages de ces thérapies anti-amyloïdes sont modestes et elles peuvent entraîner de graves effets secondaires, notamment un gonflement et des saignements cérébraux. Les porteurs d’APOE4 y sont particulièrement sujets, peut-être parce que leurs vaisseaux sanguins cérébraux sont plus chargés en amyloïdes et s’affaiblissent lorsque les plaques sont retirées.
Pourtant, les études sur l’APOE4 n’ont jamais été complètement éclipsées par la recherche sur l’amyloïde. À partir de 2018, par exemple, le neuroscientifique du Massachusetts Institute of Technology Li-Huei Tsai et ses collaborateurs ont montré comment un traitement altéré des lipides dans des types de cellules spécifiques hébergeant l’APOE4 les altère de manière à provoquer des maladies. Les neurones deviennent hyperactifs, les astrocytes et la microglie se remplissent de lipides et deviennent moins efficaces, et des cellules appelées oligodendrocytes ne parviennent pas à maintenir la gaine autour des fibres nerveuses.
Les études sur les variantes protectrices de l’APOE ont également dynamisé le domaine. En 2019, des chercheurs de la Harvard Medical School dirigés par le neuropsychologue Yakeel Quiroz ont décrit une femme colombienne qui, comme une grande partie de sa famille élargie, était porteuse d’un gène muté rare qui provoque presque inévitablement la maladie d’Alzheimer vers l’âge de 40 ans. Pourtant, contrairement à beaucoup de ses proches, elle n’a pas développé de problèmes cognitifs avant 70 ans, même si elle avait une quantité abondante d'amyloïde.
L’explication apparente était une autre mutation inhabituelle, cette fois dans ses deux copies de l’APOE3. Connue sous le nom de Christchurch pour sa découverte chez un Néo-Zélandais atteint d’une maladie lipidique en 1987, la mutation rend l’ApoE moins efficace pour se lier aux héparane sulfate protéoglycanes, des molécules qui recouvrent les cellules cérébrales – un effet qui, selon les chercheurs, pourrait expliquer les niveaux étonnamment bas de tau trouvés dans le cerveau de la femme. En juin, la même équipe a rapporté dans le New England Journal of Medicine que 27 personnes porteuses du gène Alzheimer de la famille colombienne et d’une seule copie de la mutation Christchurch sont restées en bonne santé cognitive cinq ans de plus que celles qui n’avaient pas la mutation. Ces travaux suggèrent que de futurs médicaments pourraient prévenir la maladie en imitant ce changement génétique.
Les chercheurs semblent également être parvenus à un consensus sur le débat de longue date sur la question de savoir si une thérapie doit augmenter ou bloquer l’ApoE4. L’année dernière, le directeur du National Institute on Aging, Richard Hodes, et l’ancien directeur des National Institutes of Health (NIH), Francis Collins, ont demandé à un groupe de travail de chercheurs sur l’APOE4 d’examiner les dernières preuves.
L’un des indices est venu d’études à grande échelle sur la maladie d’Alzheimer chez les Afro-Américains menées par Jeffery Vance de l’Université de Miami, qui est le mari de Pericak-Vance. Son équipe a découvert que les personnes qui ont hérité de l’APOE4 d’ancêtres africains ont des différences dans l’ADN environnant qui réduisent l’expression du gène dans les cellules cérébrales par rapport à celles qui l’ont hérité d’ancêtres européens. Cela pourrait expliquer pourquoi les porteurs afro-américains de l’APOE4 ne sont pas confrontés à un risque aussi élevé de développer la maladie d’Alzheimer que les porteurs blancs. Une autre étude menée cette année par le neurologue Michael Greicius de l’université de Stanford a trouvé deux individus blancs non hispaniques porteurs d’une copie de l’APOE4 qui sont restés sans démence et sans amyloïde à 76 et 90 ans, apparemment en raison de mutations qui ont désactivé la variante – une preuve supplémentaire que la réduction de l’ApoE4 pourrait être utile.
Après avoir examiné ces études et d’autres, le groupe de travail du NIH a publié en janvier a publié en janvier ce que Collins appelle « un article très complet » dans les Annals of Neurology. Il conclut, dit-il, que, du moins chez les personnes d’origine africaine et européenne, l’ApoE4 « est clairement une toxine ».
Ce consensus et la richesse des nouvelles découvertes font avancer les travaux sur une série de stratégies visant à cibler l’APOE4 pour traiter ou prévenir la maladie d’Alzheimer. Certaines équipes espèrent utiliser un brin d’ARN pour se lier à l’APOE4 et l’inhiber partiellement. (Une certaine quantité d’ApoE pourrait être nécessaire pour maintenir la fonction cérébrale.) De tels médicaments à base d’oligonucléotides antisens, injectés dans le liquide céphalorachidien, ont été approuvés pour traiter deux maladies neurodégénératives rares. L’équipe de Holtzman a signalé qu’un médicament antisens ciblant l’APOE pourrait freiner la production d’enchevêtrements de tau et les lésions cérébrales chez les souris atteintes de la variante APOE4. Mais il ne bloquait l’amyloïde que s’il était administré aux souris nouveau-nées.
La biochimiste Anastasia Khvorova pense que c’est parce que les souris produisaient encore trop de protéines ApoE. Son équipe de la faculté de médecine Chan de l’université du Massachusetts a rapporté plus tôt cette année que chez les souris adultes sujettes à une pathologie de type Alzheimer, de courts doubles brins de matériel génétique appelés petits ARN interférents, qui marquent l’ARN messager d’un gène pour la destruction, peuvent « faire taire l’APOE dans le cerveau de manière très puissante » sans réduire les niveaux nécessaires à l’extérieur du cerveau, explique Khvorova. Cette approche a freiné la formation de plaques amyloïdes. Les deux approches de médicaments à base d’ARN sont actuellement en cours de développement par des entreprises.
De tels médicaments seraient injectés dans le liquide céphalorachidien au moins tous les quelques mois. Mais certains chercheurs explorent ce qui serait un traitement ponctuel qui modifierait un gène dans les cellules cérébrales. À titre de preuve de concept, dans une étude de Nature Neuroscience l’automne dernier, le laboratoire de Huang a montré que des souris nouveau-nées conçues pour porter la mutation Christchurch dans l’une ou les deux copies de leur ARN messager APOE4 étaient partiellement ou totalement protégées contre le développement d’une pathologie de type Alzheimer à l’âge adulte. Un groupe dirigé par le physiologiste Lance Johnson de l'Université du Kentucky a modifié génétiquement une souris afin qu'un médicament puisse remplacer APOE4 par APOE2 dans certains types de cellules cérébrales. Lors d'une réunion l'année dernière, il a rapporté que le remplacement à mi-espérance de vie dans les seuls astrocytes avait inversé l'accumulation d'amyloïde chez les animaux.
Une approche de modification génétique a atteint le stade clinique. En 2019, un groupe dirigé par le chercheur en thérapie génique Ron Crystal de Weill Cornell Medicine a commencé à injecter un virus porteur de l’APOE2 dans la colonne vertébrale supérieure de 15 patients porteurs de deux copies de l’APOE4 et présentant une déficience cognitive légère ou des symptômes de la maladie d’Alzheimer. Des échantillons de liquide cérébral des premiers patients ont suggéré que leur cerveau produisait de l’APOE2 et produisait moins d’amyloïde et de tau, selon les comptes rendus de réunion de l’équipe de Crystal, qui prévoit de partager les résultats complets cet automne. Le sponsor de l’essai, la société de biotechnologie Lexeo Therapeutics, travaille également sur l’introduction d’un gène APOE2 avec la mutation Christchurch, et sur une autre thérapie qui ajouterait l’APOE2 et ferait taire l’APOE4.
Des vecteurs viraux plus précis en cours d’élaboration, qui ciblent le cerveau, pourraient permettre d’injecter un traitement dans le sang d’une personne plutôt que dans sa colonne vertébrale. Cependant, la modification génétique du cerveau pourrait être une mesure coûteuse et extrême pour les porteurs d’APOE4 comme Turner et Smith, qui sont cognitivement normaux mais présentent un risque élevé de développer la maladie d’Alzheimer.
Holtzman et son groupe ont développé une intervention moins radicale : un anticorps qui se lie à une forme particulièrement nocive de la protéine ApoE4 et l’élimine. Ils ont montré qu’il peut prévenir une pathologie de type Alzheimer chez la souris. Le groupe de Tsai étudie si un cocktail de médicaments ou de suppléments déjà approuvés pourrait contrecarrer certains des dommages causés par l’APOE4 que son groupe a observés. L’équipe a signalé, par exemple, que la choline, un nutriment, peut corriger le traitement des lipides dans des astrocytes humains cultivés contenant de l’APOE4. Dans le cadre d’un petit essai au Texas, les collaborateurs de Tsai testent les effets des suppléments de choline sur les profils lipidiques du liquide céphalorachidien de personnes cognitivement normales possédant au moins une copie de l’APOE4.
« C’est un petit pas vers la réalisation de notre idée d’un moyen abordable, accessible et sûr de faire avancer les choses pour les porteurs », déclare Tsai.
TOUS CES PROGRÈS suscitent l’espoir chez les personnes qui savent qu’elles sont porteuses de la variante APOE4 et qui sont désireuses d’améliorer leurs chances. Leurs rangs s’agrandissent rapidement. Beaucoup, y compris moi, apprennent leur statut grâce à des tests ADN accessibles directement aux consommateurs tels que 23andMe. Plusieurs médecins-chercheurs interrogés pour cet article ont déclaré que les porteurs d’APOE4 se présentent à leurs cabinets, inquiets du risque qu’ils courent.
Un traitement que quelqu’un comme moi pourrait prendre pour se prémunir contre la maladie d’Alzheimer semble désormais possible, même si cela pourrait prendre des années. Que faisons-nous, nous les porteurs d’APOE4, en attendant ? En tant que porteur d’une copie d’APOE3 et d’une copie d’APOE4, j’ai trois fois plus de risques qu’une personne blanche porteuse de deux copies d’APOE3 – une chance de 30 %, ou plus selon certaines estimations, de développer la maladie d’Alzheimer à l’âge de 85 ans, soit dans 26 ans. J’ai hérité de l’APOE4 de mon père, qui souffrait de démence et qui se souvenait à peine de moi lorsqu’il est décédé.
Hussein Yassine, chercheur sur la maladie d’Alzheimer à l’Université de Californie du Sud (USC), souligne que le fait d’être porteur de l’APOE4 n’est pas intrinsèquement un défaut. La variante pourrait en fait être la forme originale du gène, celle que possédaient nos premiers ancêtres humains, et pourrait en fait offrir des avantages. « Il y a une raison pour laquelle tant de personnes en sont porteuses », dit-il. Certaines études ont suggéré que l’APOE4 aide les porteurs à lutter contre les agents pathogènes de l’enfance. D’autres recherches suggèrent même que les jeunes adultes porteurs de l’APOE4 ont une meilleure mémoire spatiale et d’autres avantages cognitifs subtils.
Mais il existe également des preuves que le mode de vie sédentaire et la forte consommation de sucres raffinés courants chez les humains modernes pourraient exacerber les inconvénients du gène, dit Yassine. « La vie moderne ne convient pas à certaines personnes porteuses de l’APOE4. »
Les conseils des scientifiques aux porteurs de l’APOE4 : bien manger, faire de l’exercice, dormir suffisamment, contrôler le cholestérol et la tension artérielle et stimuler son esprit. Toutes ces habitudes semblent réduire le risque de maladie d’Alzheimer pour tout le monde. Au Centre de santé cérébrale personnalisée de l’USC, lancé en 2023 expressément pour recruter des porteurs d’APOE4 pour des études de prévention et de découverte de médicaments, Yassine prévoit des essais pour tester certaines de ces stratégies de style de vie. Il termine également une étude sur des doses élevées d’oméga-3, une forme de graisse qui, selon certaines données, protège les porteurs d’APOE4 de la neuroinflammation.
Turner est resté actif en aidant à recruter des Afro-Américains pour l’essai de prévention, en partie grâce à des visites dans les églises locales. Smith garde l’esprit vif en tant qu’arbitre de matchs de softball, qui ont « beaucoup de règles », dit-il. Les deux se réconfortent dans le fait que s’ils présentent des signes de déficience cognitive, éventuellement pendant qu’ils reçoivent un placebo, ils peuvent choisir de recevoir du lécanemab. « C’est ce qui est important pour moi, que vous ayez le choix de recevoir le médicament », dit Smith.
Quant à moi, je me suis inscrit au même essai, mais j’ai appris que je n’avais pas assez d’amyloïde pour m’inscrire. Donc, comme beaucoup d’autres APOE4 – il existe un forum en ligne (APOE4.info) où ils se réunissent – je parcours maintenant le Web à la recherche de nouvelles recherches sur la variante, je m’essaie à prendre des suppléments comme la choline et les oméga-3, et je réfléchis à suivre mes résultats aux tests cognitifs en ligne. J’espère que le déclin éventuel sera suffisamment lent pour que je puisse aider mes deux filles, qui, selon 23andMe, ne sont pas porteuses du gène APOE4, à faire des projets pour moi.
Et j’essaie de me rappeler ce que les experts m’ont dit : de nombreuses personnes porteuses d’un seul gène APOE4 ne développeront pas de problèmes cognitifs, et même certaines personnes porteuses de deux copies ne développeront pas de démence à 90 ans. « Il faut comprendre que le fait d’être porteur n’est pas un diagnostic », explique Goldie Smith Byrd, chercheuse sur la maladie d’Alzheimer à la Wake Forest University School of Medicine, qui a fondé un centre de recherche en santé communautaire qui soutient les Noirs américains atteints de la maladie d’Alzheimer dans leur famille. Ses mots résonnent en moi : « Mettez de l’ordre dans vos affaires, essayez d’être en aussi bonne santé que possible. Et essayez de ne pas trop vous stresser à ce sujet. »